Shule Fan , Zeyi Wan , Yuhua Qu , Wenxia Lu, Xiangzhi Li, Feifei Yang, Hua Zhang
{"title":"设计和优化新型四氢-β-咔啉基 HDAC 抑制剂,使其具有抑制肿瘤细胞生长和转移的强效活性","authors":"Shule Fan , Zeyi Wan , Yuhua Qu , Wenxia Lu, Xiangzhi Li, Feifei Yang, Hua Zhang","doi":"10.1016/j.bmcl.2024.129986","DOIUrl":null,"url":null,"abstract":"<div><div>Histone deacetylases (HDACs) are validated drug targets for various therapeutic applications. A series of Tetrahydro-β-carboline-based hydroxamate derivatives, designed as HDAC inhibitors (HDACis), were synthesized. Compound <strong>11g</strong> exhibited strong inhibitory activity against HDAC1 and the A549 cancer cell line. Additionally, this compound increased the levels of acetylated histone H3 and H4. Notably, <strong>11g</strong> effectively arrested A549 cells in the G2/M phase and also increased ROS production and DNA damage, thereby inducing apoptosis. Further molecular docking experiments illustrated the potential interactions between compound <strong>11g</strong> and HDAC1. These findings suggested that the novel Tetrahydro-β-carboline-based HDACis could serve as a promising framework for further optimization as anticancer agents.</div></div>","PeriodicalId":256,"journal":{"name":"Bioorganic & Medicinal Chemistry Letters","volume":"114 ","pages":"Article 129986"},"PeriodicalIF":2.2000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design and optimization of novel Tetrahydro-β-carboline-based HDAC inhibitors with potent activities against tumor cell growth and metastasis\",\"authors\":\"Shule Fan , Zeyi Wan , Yuhua Qu , Wenxia Lu, Xiangzhi Li, Feifei Yang, Hua Zhang\",\"doi\":\"10.1016/j.bmcl.2024.129986\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Histone deacetylases (HDACs) are validated drug targets for various therapeutic applications. A series of Tetrahydro-β-carboline-based hydroxamate derivatives, designed as HDAC inhibitors (HDACis), were synthesized. Compound <strong>11g</strong> exhibited strong inhibitory activity against HDAC1 and the A549 cancer cell line. Additionally, this compound increased the levels of acetylated histone H3 and H4. Notably, <strong>11g</strong> effectively arrested A549 cells in the G2/M phase and also increased ROS production and DNA damage, thereby inducing apoptosis. Further molecular docking experiments illustrated the potential interactions between compound <strong>11g</strong> and HDAC1. These findings suggested that the novel Tetrahydro-β-carboline-based HDACis could serve as a promising framework for further optimization as anticancer agents.</div></div>\",\"PeriodicalId\":256,\"journal\":{\"name\":\"Bioorganic & Medicinal Chemistry Letters\",\"volume\":\"114 \",\"pages\":\"Article 129986\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioorganic & Medicinal Chemistry Letters\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0960894X24003883\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/10 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic & Medicinal Chemistry Letters","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0960894X24003883","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/10 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Design and optimization of novel Tetrahydro-β-carboline-based HDAC inhibitors with potent activities against tumor cell growth and metastasis
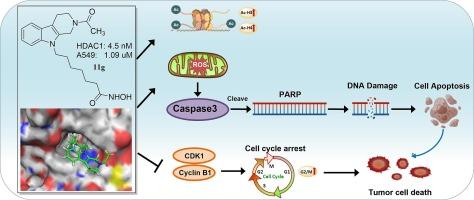
Histone deacetylases (HDACs) are validated drug targets for various therapeutic applications. A series of Tetrahydro-β-carboline-based hydroxamate derivatives, designed as HDAC inhibitors (HDACis), were synthesized. Compound 11g exhibited strong inhibitory activity against HDAC1 and the A549 cancer cell line. Additionally, this compound increased the levels of acetylated histone H3 and H4. Notably, 11g effectively arrested A549 cells in the G2/M phase and also increased ROS production and DNA damage, thereby inducing apoptosis. Further molecular docking experiments illustrated the potential interactions between compound 11g and HDAC1. These findings suggested that the novel Tetrahydro-β-carboline-based HDACis could serve as a promising framework for further optimization as anticancer agents.
期刊介绍:
Bioorganic & Medicinal Chemistry Letters presents preliminary experimental or theoretical research results of outstanding significance and timeliness on all aspects of science at the interface of chemistry and biology and on major advances in drug design and development. The journal publishes articles in the form of communications reporting experimental or theoretical results of special interest, and strives to provide maximum dissemination to a large, international audience.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
