{"title":"KMT2C/D 缺乏通过调节 KDM6A 介导的表观遗传重塑促进乳腺癌转移","authors":"Zhao Huang, Wei Zhao","doi":"10.1002/mog2.95","DOIUrl":null,"url":null,"abstract":"<p>On June 26, 2024, Kornelia Polyak's team published an article in Nature Cell Biology that revealed a new mechanism by which histone-lysine N-methyltransferase 2 C (<i>KMT2C)</i> or <i>KMT2D</i> deletion promotes metalloproteinase 3 (MMP3) expression in a lysine-specific demethylase 6 A (KDM6A)-dependent manner, thereby driving brain metastasis (BMs) of triple-negative breast cancer (TNBC). It provides a new perspective for the treatment of TNBC at the epigenetic level.<span><sup>1</sup></span> Breast cancer is the most common neoplasm and the leading cause of tumor-related death in women worldwide. Breast cancer subtypes can be classified according to the expression of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2.<span><sup>2</sup></span> TNBC is characterized by a high level of cell invasiveness and visceral metastasis to organs, usually brain, lungs, and liver, with an average survival time of 18 months. Although TNBC accounts for only about 15%–20% of all breast cancers, it is the subtype with the worst prognosis of breast cancer. Before the advent of immunotherapy, systemic chemotherapies including taxanes, anthracyclines and/or platinum were the predominant first-line treatment options for TNBC. However, the median overall survival of metastatic TNBC is only 9–12 months, and the 5-year survival rate is approximately 12%, which is a serious unmet medical need.<span><sup>3</sup></span></p><p>The most commonly mutated gene in BMs of breast cancer has been reported to be <i>KMT2C/D</i>, and the results of gene sequencing showed that <i>KMT2C</i> and <i>KMT2D</i> were significantly reduced in distal metastasis compared to the primary tumor, suggesting that functional loss of <i>KMT2C/D</i> plays a role in BMs of breast cancer. <i>KMT2C</i> and <i>KMT2D</i> catalyze the methylation of unmethylated H3K4 sites to form H3K4me1, which can be enriched in active enhancer and promoter regions. This modification recruits other histone modifying enzymes, including histone H3K27 acetyltransferases (such as P300) and demethylases (such as KDM6A), which are essential for the regulation of gene expression. As the core components of the SET1-associated protein epigenetic regulatory complex (COMPASS), <i>KMT2C</i> and <i>KMT2D</i> profoundly regulate the epigenetic landscape.<span><sup>4</sup></span> However, it is still unclear how <i>KMT2C</i>/<i>D</i> mutations affect the epigenetic and transcriptomic landscape to promote tumorigenesis.</p><p>Recently, Marco Seehawer and colleagues from Dana-Farber Cancer Institute in the United States found that the loss of <i>KMT2C</i> or <i>KMT2D</i> can lead to metastasis (especially BMs) in a nonmetastatic TNBC mouse model. Mechanistic studies revealed that the loss of <i>KMT2C</i> or <i>KMT2D</i> promotes the expression of matrix MMP3 in a KDM6A-dependent manner, thereby driving BMs of TNBC.<span><sup>1</sup></span> This study provides not only a new perspective for the treatment of TNBC at the epigenetic level, but also a potential target for the development of targeted therapeutic strategies for BMs.</p><p>First, Seehawer et al. showed that the expression level of <i>KMT2C</i> and <i>KMT2D</i> were significantly reduced in distal metastasis compared with primary breast tumors. To investigate the role of <i>KMT2C/D</i> deletion in BMs, they injected the nonmetastatic mouse breast tumor cell line 168FARN tagged with H2B-mCherry into the mammary fat pad (MFP) of mice and found that while orthotopic tumor growth was unaffected, mCherry+ micrometastatic lesions appeared in the lung, liver, brain in mice injected with <i>KMT2C</i> or <i>KMT2D</i> knockout (KO) 168FARN cells. This suggests that they are indeed derived from KO cells.</p><p>Subsequently, the researchers performed single-cell RNA sequencing (scRNA-seq) on 168FARN-derived primary tumors injected with MFP. Differentially expressed gene analysis found that <i>Ly6a</i>, <i>Bst2</i>, <i>Ifi27l2a</i>, and <i>Stat1</i> expression were significantly upregulated in KO tumor cells, suggesting a proinflammatory environment generated by potential activation of interferon signaling. In addition, they found that the loss of <i>KMT2C</i> and <i>KMT2D</i> had specific effects on different types of cell subsets in the tumor microenvironment.</p><p>To dissect the cell-intrinsic changes caused by the loss of <i>KMT2C</i> or <i>KMT2D</i>, Seehawer et al. focused on histones that are direct targets of <i>KMT2C/D</i>. Using quantitative histone mass spectrometry, immunoblotting and chromatin immunoprecipitation sequencing (ChIP-seq) approaches, they found locus-specific but not global changes of H3K4me1 in both KO cells. By contrast, H3K27me3 was altered globally instead of a locus-specific manner. KDM6A, as an H3K27me3 demethylase, is one of the components of COMPASS complex. KDM6A-ChIP-seq results showed that the peaks in KO cells increased significantly.</p><p>Subsequently, Seehawer et al. performed RNA-seq analysis and found that the loss of <i>KMT2C</i> or <i>KMT2D</i> induced the epithelial-mesenchymal hybriditic transformation (EMT) state, altering the EMT balance and thus promoting metastasis. It is worth mentioning that the role of <i>KMT2C/D</i> deletion in promoting metastasis was observed even in the absence of EMT. Further integrating RNA-seq and ChIP-seq data, they found that <i>KMT2C/D</i> deficiency can regulate gene expression by affecting chromatin state, which may affect tumor metastasis, and MMP3KDM6A may be a mediator of transcriptome changes associated with <i>KMT2C/D</i> deficiency.</p><p>To identify common drivers inducing the epigenetic events and the metastatic phenotype of <i>KMT2C</i>/<i>D</i> KO cells, Seehawer et al. integrated data from ChIP-seq, RNA-seq and scRNA-seq, combined with experimental validation, confirming that <i>MMP3</i> was the only overlapping gene. More importantly, analysis of clinical samples found that <i>MMP3</i> was overexpressed in <i>KMT2C</i>-mutated TNBCs compared with WT tumors, and the frequency of <i>KMT2C</i> mutations was significantly higher in <i>MMP3</i>-high-expressing tumors. Further, downregulation of <i>MMP3</i> by shRNA significantly reduced BMs of <i>KMT2/D</i> KO cells in a mouse model. Existing clinical trials of MMP inhibitors have shown high toxicity and low efficacy. Fortunately, Seehawer et al. found that inhibiting <i>KDM6A</i> by shRNA or pharmacology could reduce the expression of <i>MMP3</i> and inhibit BMs of <i>KMT2C/D</i> KO cells in vivo. In addition, they also found that KDM6A inhibition was tolerable in mice, suggesting that KDM6A inhibitors may have potential clinical application value for TNBC patients (Figure 1).</p><p>Epigenetic regulation is widely considered as a major tumor-suppressive mechanism for many cancer types, including breast cancer.<span><sup>5</sup></span> In summary, this study elucidated the molecular mechanism of chromatin remodeling and histone modification changes caused by <i>KMT2C/D</i> mutations, which indirectly affected the expression of MMP3 through KDM5A, thereby inducing TNBC distant metastasis. Inhibiting the activity of KDM6A can lead to the suppression of MMP3, which represses the metastasis caused by <i>KMT2C/D</i> mutations in TNBC. The results of this study increase the understanding of the mechanism of TNBC BMs, provide a potential target for the development of targeted therapeutic strategies for BMs. As KDM6A inhibitors have not yet been used in clinical treatment for cancers, this study provided valuable information for the development of novel KDM6A-based therapeutic strategies for the treatment of TNBC.</p><p>Wei Zhao provided acquisition, analysis, and interpretation of data. Zhao Huang was responsible for writing, reviewing, and revising the paper. Both authors have read and approved the final manuscript.</p><p>Not applicable.</p><p>Not applicable.</p>","PeriodicalId":100902,"journal":{"name":"MedComm – Oncology","volume":"3 4","pages":""},"PeriodicalIF":2.2000,"publicationDate":"2024-10-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mog2.95","citationCount":"0","resultStr":"{\"title\":\"KMT2C/D deficiency promotes breast cancer metastasis by regulating KDM6A-mediated epigenetic remodeling\",\"authors\":\"Zhao Huang, Wei Zhao\",\"doi\":\"10.1002/mog2.95\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>On June 26, 2024, Kornelia Polyak's team published an article in Nature Cell Biology that revealed a new mechanism by which histone-lysine N-methyltransferase 2 C (<i>KMT2C)</i> or <i>KMT2D</i> deletion promotes metalloproteinase 3 (MMP3) expression in a lysine-specific demethylase 6 A (KDM6A)-dependent manner, thereby driving brain metastasis (BMs) of triple-negative breast cancer (TNBC). It provides a new perspective for the treatment of TNBC at the epigenetic level.<span><sup>1</sup></span> Breast cancer is the most common neoplasm and the leading cause of tumor-related death in women worldwide. Breast cancer subtypes can be classified according to the expression of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2.<span><sup>2</sup></span> TNBC is characterized by a high level of cell invasiveness and visceral metastasis to organs, usually brain, lungs, and liver, with an average survival time of 18 months. Although TNBC accounts for only about 15%–20% of all breast cancers, it is the subtype with the worst prognosis of breast cancer. Before the advent of immunotherapy, systemic chemotherapies including taxanes, anthracyclines and/or platinum were the predominant first-line treatment options for TNBC. However, the median overall survival of metastatic TNBC is only 9–12 months, and the 5-year survival rate is approximately 12%, which is a serious unmet medical need.<span><sup>3</sup></span></p><p>The most commonly mutated gene in BMs of breast cancer has been reported to be <i>KMT2C/D</i>, and the results of gene sequencing showed that <i>KMT2C</i> and <i>KMT2D</i> were significantly reduced in distal metastasis compared to the primary tumor, suggesting that functional loss of <i>KMT2C/D</i> plays a role in BMs of breast cancer. <i>KMT2C</i> and <i>KMT2D</i> catalyze the methylation of unmethylated H3K4 sites to form H3K4me1, which can be enriched in active enhancer and promoter regions. This modification recruits other histone modifying enzymes, including histone H3K27 acetyltransferases (such as P300) and demethylases (such as KDM6A), which are essential for the regulation of gene expression. As the core components of the SET1-associated protein epigenetic regulatory complex (COMPASS), <i>KMT2C</i> and <i>KMT2D</i> profoundly regulate the epigenetic landscape.<span><sup>4</sup></span> However, it is still unclear how <i>KMT2C</i>/<i>D</i> mutations affect the epigenetic and transcriptomic landscape to promote tumorigenesis.</p><p>Recently, Marco Seehawer and colleagues from Dana-Farber Cancer Institute in the United States found that the loss of <i>KMT2C</i> or <i>KMT2D</i> can lead to metastasis (especially BMs) in a nonmetastatic TNBC mouse model. Mechanistic studies revealed that the loss of <i>KMT2C</i> or <i>KMT2D</i> promotes the expression of matrix MMP3 in a KDM6A-dependent manner, thereby driving BMs of TNBC.<span><sup>1</sup></span> This study provides not only a new perspective for the treatment of TNBC at the epigenetic level, but also a potential target for the development of targeted therapeutic strategies for BMs.</p><p>First, Seehawer et al. showed that the expression level of <i>KMT2C</i> and <i>KMT2D</i> were significantly reduced in distal metastasis compared with primary breast tumors. To investigate the role of <i>KMT2C/D</i> deletion in BMs, they injected the nonmetastatic mouse breast tumor cell line 168FARN tagged with H2B-mCherry into the mammary fat pad (MFP) of mice and found that while orthotopic tumor growth was unaffected, mCherry+ micrometastatic lesions appeared in the lung, liver, brain in mice injected with <i>KMT2C</i> or <i>KMT2D</i> knockout (KO) 168FARN cells. This suggests that they are indeed derived from KO cells.</p><p>Subsequently, the researchers performed single-cell RNA sequencing (scRNA-seq) on 168FARN-derived primary tumors injected with MFP. Differentially expressed gene analysis found that <i>Ly6a</i>, <i>Bst2</i>, <i>Ifi27l2a</i>, and <i>Stat1</i> expression were significantly upregulated in KO tumor cells, suggesting a proinflammatory environment generated by potential activation of interferon signaling. In addition, they found that the loss of <i>KMT2C</i> and <i>KMT2D</i> had specific effects on different types of cell subsets in the tumor microenvironment.</p><p>To dissect the cell-intrinsic changes caused by the loss of <i>KMT2C</i> or <i>KMT2D</i>, Seehawer et al. focused on histones that are direct targets of <i>KMT2C/D</i>. Using quantitative histone mass spectrometry, immunoblotting and chromatin immunoprecipitation sequencing (ChIP-seq) approaches, they found locus-specific but not global changes of H3K4me1 in both KO cells. By contrast, H3K27me3 was altered globally instead of a locus-specific manner. KDM6A, as an H3K27me3 demethylase, is one of the components of COMPASS complex. KDM6A-ChIP-seq results showed that the peaks in KO cells increased significantly.</p><p>Subsequently, Seehawer et al. performed RNA-seq analysis and found that the loss of <i>KMT2C</i> or <i>KMT2D</i> induced the epithelial-mesenchymal hybriditic transformation (EMT) state, altering the EMT balance and thus promoting metastasis. It is worth mentioning that the role of <i>KMT2C/D</i> deletion in promoting metastasis was observed even in the absence of EMT. Further integrating RNA-seq and ChIP-seq data, they found that <i>KMT2C/D</i> deficiency can regulate gene expression by affecting chromatin state, which may affect tumor metastasis, and MMP3KDM6A may be a mediator of transcriptome changes associated with <i>KMT2C/D</i> deficiency.</p><p>To identify common drivers inducing the epigenetic events and the metastatic phenotype of <i>KMT2C</i>/<i>D</i> KO cells, Seehawer et al. integrated data from ChIP-seq, RNA-seq and scRNA-seq, combined with experimental validation, confirming that <i>MMP3</i> was the only overlapping gene. More importantly, analysis of clinical samples found that <i>MMP3</i> was overexpressed in <i>KMT2C</i>-mutated TNBCs compared with WT tumors, and the frequency of <i>KMT2C</i> mutations was significantly higher in <i>MMP3</i>-high-expressing tumors. Further, downregulation of <i>MMP3</i> by shRNA significantly reduced BMs of <i>KMT2/D</i> KO cells in a mouse model. Existing clinical trials of MMP inhibitors have shown high toxicity and low efficacy. Fortunately, Seehawer et al. found that inhibiting <i>KDM6A</i> by shRNA or pharmacology could reduce the expression of <i>MMP3</i> and inhibit BMs of <i>KMT2C/D</i> KO cells in vivo. In addition, they also found that KDM6A inhibition was tolerable in mice, suggesting that KDM6A inhibitors may have potential clinical application value for TNBC patients (Figure 1).</p><p>Epigenetic regulation is widely considered as a major tumor-suppressive mechanism for many cancer types, including breast cancer.<span><sup>5</sup></span> In summary, this study elucidated the molecular mechanism of chromatin remodeling and histone modification changes caused by <i>KMT2C/D</i> mutations, which indirectly affected the expression of MMP3 through KDM5A, thereby inducing TNBC distant metastasis. Inhibiting the activity of KDM6A can lead to the suppression of MMP3, which represses the metastasis caused by <i>KMT2C/D</i> mutations in TNBC. The results of this study increase the understanding of the mechanism of TNBC BMs, provide a potential target for the development of targeted therapeutic strategies for BMs. As KDM6A inhibitors have not yet been used in clinical treatment for cancers, this study provided valuable information for the development of novel KDM6A-based therapeutic strategies for the treatment of TNBC.</p><p>Wei Zhao provided acquisition, analysis, and interpretation of data. Zhao Huang was responsible for writing, reviewing, and revising the paper. Both authors have read and approved the final manuscript.</p><p>Not applicable.</p><p>Not applicable.</p>\",\"PeriodicalId\":100902,\"journal\":{\"name\":\"MedComm – Oncology\",\"volume\":\"3 4\",\"pages\":\"\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-10-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mog2.95\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"MedComm – Oncology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mog2.95\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm – Oncology","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mog2.95","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
KMT2C/D deficiency promotes breast cancer metastasis by regulating KDM6A-mediated epigenetic remodeling
On June 26, 2024, Kornelia Polyak's team published an article in Nature Cell Biology that revealed a new mechanism by which histone-lysine N-methyltransferase 2 C (KMT2C) or KMT2D deletion promotes metalloproteinase 3 (MMP3) expression in a lysine-specific demethylase 6 A (KDM6A)-dependent manner, thereby driving brain metastasis (BMs) of triple-negative breast cancer (TNBC). It provides a new perspective for the treatment of TNBC at the epigenetic level.1 Breast cancer is the most common neoplasm and the leading cause of tumor-related death in women worldwide. Breast cancer subtypes can be classified according to the expression of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2.2 TNBC is characterized by a high level of cell invasiveness and visceral metastasis to organs, usually brain, lungs, and liver, with an average survival time of 18 months. Although TNBC accounts for only about 15%–20% of all breast cancers, it is the subtype with the worst prognosis of breast cancer. Before the advent of immunotherapy, systemic chemotherapies including taxanes, anthracyclines and/or platinum were the predominant first-line treatment options for TNBC. However, the median overall survival of metastatic TNBC is only 9–12 months, and the 5-year survival rate is approximately 12%, which is a serious unmet medical need.3
The most commonly mutated gene in BMs of breast cancer has been reported to be KMT2C/D, and the results of gene sequencing showed that KMT2C and KMT2D were significantly reduced in distal metastasis compared to the primary tumor, suggesting that functional loss of KMT2C/D plays a role in BMs of breast cancer. KMT2C and KMT2D catalyze the methylation of unmethylated H3K4 sites to form H3K4me1, which can be enriched in active enhancer and promoter regions. This modification recruits other histone modifying enzymes, including histone H3K27 acetyltransferases (such as P300) and demethylases (such as KDM6A), which are essential for the regulation of gene expression. As the core components of the SET1-associated protein epigenetic regulatory complex (COMPASS), KMT2C and KMT2D profoundly regulate the epigenetic landscape.4 However, it is still unclear how KMT2C/D mutations affect the epigenetic and transcriptomic landscape to promote tumorigenesis.
Recently, Marco Seehawer and colleagues from Dana-Farber Cancer Institute in the United States found that the loss of KMT2C or KMT2D can lead to metastasis (especially BMs) in a nonmetastatic TNBC mouse model. Mechanistic studies revealed that the loss of KMT2C or KMT2D promotes the expression of matrix MMP3 in a KDM6A-dependent manner, thereby driving BMs of TNBC.1 This study provides not only a new perspective for the treatment of TNBC at the epigenetic level, but also a potential target for the development of targeted therapeutic strategies for BMs.
First, Seehawer et al. showed that the expression level of KMT2C and KMT2D were significantly reduced in distal metastasis compared with primary breast tumors. To investigate the role of KMT2C/D deletion in BMs, they injected the nonmetastatic mouse breast tumor cell line 168FARN tagged with H2B-mCherry into the mammary fat pad (MFP) of mice and found that while orthotopic tumor growth was unaffected, mCherry+ micrometastatic lesions appeared in the lung, liver, brain in mice injected with KMT2C or KMT2D knockout (KO) 168FARN cells. This suggests that they are indeed derived from KO cells.
Subsequently, the researchers performed single-cell RNA sequencing (scRNA-seq) on 168FARN-derived primary tumors injected with MFP. Differentially expressed gene analysis found that Ly6a, Bst2, Ifi27l2a, and Stat1 expression were significantly upregulated in KO tumor cells, suggesting a proinflammatory environment generated by potential activation of interferon signaling. In addition, they found that the loss of KMT2C and KMT2D had specific effects on different types of cell subsets in the tumor microenvironment.
To dissect the cell-intrinsic changes caused by the loss of KMT2C or KMT2D, Seehawer et al. focused on histones that are direct targets of KMT2C/D. Using quantitative histone mass spectrometry, immunoblotting and chromatin immunoprecipitation sequencing (ChIP-seq) approaches, they found locus-specific but not global changes of H3K4me1 in both KO cells. By contrast, H3K27me3 was altered globally instead of a locus-specific manner. KDM6A, as an H3K27me3 demethylase, is one of the components of COMPASS complex. KDM6A-ChIP-seq results showed that the peaks in KO cells increased significantly.
Subsequently, Seehawer et al. performed RNA-seq analysis and found that the loss of KMT2C or KMT2D induced the epithelial-mesenchymal hybriditic transformation (EMT) state, altering the EMT balance and thus promoting metastasis. It is worth mentioning that the role of KMT2C/D deletion in promoting metastasis was observed even in the absence of EMT. Further integrating RNA-seq and ChIP-seq data, they found that KMT2C/D deficiency can regulate gene expression by affecting chromatin state, which may affect tumor metastasis, and MMP3KDM6A may be a mediator of transcriptome changes associated with KMT2C/D deficiency.
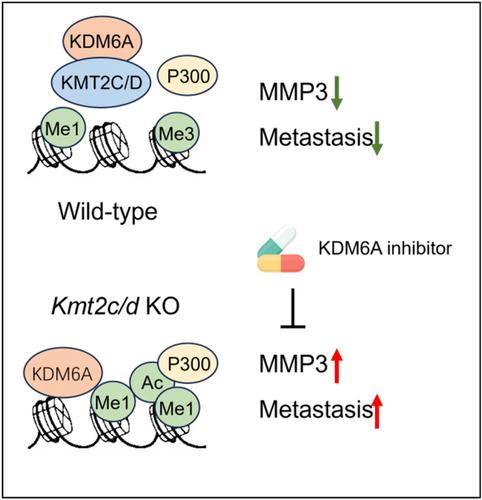
To identify common drivers inducing the epigenetic events and the metastatic phenotype of KMT2C/D KO cells, Seehawer et al. integrated data from ChIP-seq, RNA-seq and scRNA-seq, combined with experimental validation, confirming that MMP3 was the only overlapping gene. More importantly, analysis of clinical samples found that MMP3 was overexpressed in KMT2C-mutated TNBCs compared with WT tumors, and the frequency of KMT2C mutations was significantly higher in MMP3-high-expressing tumors. Further, downregulation of MMP3 by shRNA significantly reduced BMs of KMT2/D KO cells in a mouse model. Existing clinical trials of MMP inhibitors have shown high toxicity and low efficacy. Fortunately, Seehawer et al. found that inhibiting KDM6A by shRNA or pharmacology could reduce the expression of MMP3 and inhibit BMs of KMT2C/D KO cells in vivo. In addition, they also found that KDM6A inhibition was tolerable in mice, suggesting that KDM6A inhibitors may have potential clinical application value for TNBC patients (Figure 1).
Epigenetic regulation is widely considered as a major tumor-suppressive mechanism for many cancer types, including breast cancer.5 In summary, this study elucidated the molecular mechanism of chromatin remodeling and histone modification changes caused by KMT2C/D mutations, which indirectly affected the expression of MMP3 through KDM5A, thereby inducing TNBC distant metastasis. Inhibiting the activity of KDM6A can lead to the suppression of MMP3, which represses the metastasis caused by KMT2C/D mutations in TNBC. The results of this study increase the understanding of the mechanism of TNBC BMs, provide a potential target for the development of targeted therapeutic strategies for BMs. As KDM6A inhibitors have not yet been used in clinical treatment for cancers, this study provided valuable information for the development of novel KDM6A-based therapeutic strategies for the treatment of TNBC.
Wei Zhao provided acquisition, analysis, and interpretation of data. Zhao Huang was responsible for writing, reviewing, and revising the paper. Both authors have read and approved the final manuscript.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
