{"title":"电致变色六氰钌酸铁电化学中的 H/D 同位素效应","authors":"Lena Gerhards, Izabella Brand, Gunther Wittstock","doi":"10.1002/celc.202300824","DOIUrl":null,"url":null,"abstract":"<p>The electrochemical deposition of iron hexacyanoruthenate (Fe−HCR) on gold electrodes was studied in electrolytes prepared with light and heavy water. Cyclic voltammetry of the material during preparation and after transfer to a precursor-free solution exhibits two reductions peaks in H<sub>2</sub>O-based electrolytes but only one reduction peak in D<sub>2</sub>O-based electrolytes. The voltammetric behavior changes reversibly upon transfer of the material between D<sub>2</sub>O-based and H<sub>2</sub>O-based 1 mol L<sup>−1</sup> KCl solutions. No clear structural differences between samples prepared in D<sub>2</sub>O and H<sub>2</sub>O were detected by means of X-ray photoelectron spectroscopy (XPS) and polarization modulation infrared reflection absorption spectroscopy (PM IRRAS). We noted a relatively slow exchange of coordinated water and a fast exchange of zeolitic water. Using voltammetric experiments we could rule out simple effects of solution conductivity for K<sup>+</sup>, participation of H<sup>+</sup>/D<sup>+</sup> in the charge compensation and surface effects on the observed dependence of the peak splitting on the isotopic composition of the solvent. The most likely reason for the observed behavior is the different structure of the H-bonded water network of coordinated H<sub>2</sub>O and zeolitic H<sub>2</sub>O/D<sub>2</sub>O which is supported by the PM IRRAS data.</p>","PeriodicalId":142,"journal":{"name":"ChemElectroChem","volume":"11 20","pages":""},"PeriodicalIF":5.2000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/celc.202300824","citationCount":"0","resultStr":"{\"title\":\"H/D Isotope Effects in the Electrochemistry of Electrochromic Iron Hexacyanoruthenate\",\"authors\":\"Lena Gerhards, Izabella Brand, Gunther Wittstock\",\"doi\":\"10.1002/celc.202300824\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The electrochemical deposition of iron hexacyanoruthenate (Fe−HCR) on gold electrodes was studied in electrolytes prepared with light and heavy water. Cyclic voltammetry of the material during preparation and after transfer to a precursor-free solution exhibits two reductions peaks in H<sub>2</sub>O-based electrolytes but only one reduction peak in D<sub>2</sub>O-based electrolytes. The voltammetric behavior changes reversibly upon transfer of the material between D<sub>2</sub>O-based and H<sub>2</sub>O-based 1 mol L<sup>−1</sup> KCl solutions. No clear structural differences between samples prepared in D<sub>2</sub>O and H<sub>2</sub>O were detected by means of X-ray photoelectron spectroscopy (XPS) and polarization modulation infrared reflection absorption spectroscopy (PM IRRAS). We noted a relatively slow exchange of coordinated water and a fast exchange of zeolitic water. Using voltammetric experiments we could rule out simple effects of solution conductivity for K<sup>+</sup>, participation of H<sup>+</sup>/D<sup>+</sup> in the charge compensation and surface effects on the observed dependence of the peak splitting on the isotopic composition of the solvent. The most likely reason for the observed behavior is the different structure of the H-bonded water network of coordinated H<sub>2</sub>O and zeolitic H<sub>2</sub>O/D<sub>2</sub>O which is supported by the PM IRRAS data.</p>\",\"PeriodicalId\":142,\"journal\":{\"name\":\"ChemElectroChem\",\"volume\":\"11 20\",\"pages\":\"\"},\"PeriodicalIF\":5.2000,\"publicationDate\":\"2024-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/celc.202300824\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ChemElectroChem\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/celc.202300824\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"ELECTROCHEMISTRY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemElectroChem","FirstCategoryId":"92","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/celc.202300824","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ELECTROCHEMISTRY","Score":null,"Total":0}
H/D Isotope Effects in the Electrochemistry of Electrochromic Iron Hexacyanoruthenate
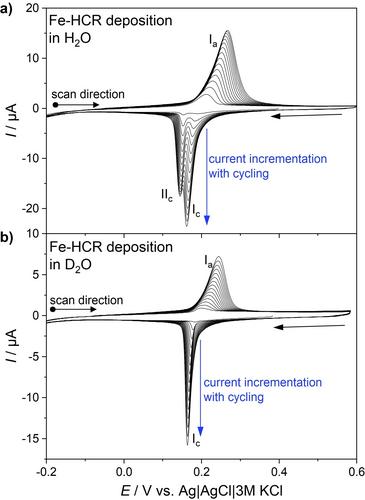
The electrochemical deposition of iron hexacyanoruthenate (Fe−HCR) on gold electrodes was studied in electrolytes prepared with light and heavy water. Cyclic voltammetry of the material during preparation and after transfer to a precursor-free solution exhibits two reductions peaks in H2O-based electrolytes but only one reduction peak in D2O-based electrolytes. The voltammetric behavior changes reversibly upon transfer of the material between D2O-based and H2O-based 1 mol L−1 KCl solutions. No clear structural differences between samples prepared in D2O and H2O were detected by means of X-ray photoelectron spectroscopy (XPS) and polarization modulation infrared reflection absorption spectroscopy (PM IRRAS). We noted a relatively slow exchange of coordinated water and a fast exchange of zeolitic water. Using voltammetric experiments we could rule out simple effects of solution conductivity for K+, participation of H+/D+ in the charge compensation and surface effects on the observed dependence of the peak splitting on the isotopic composition of the solvent. The most likely reason for the observed behavior is the different structure of the H-bonded water network of coordinated H2O and zeolitic H2O/D2O which is supported by the PM IRRAS data.
期刊介绍:
ChemElectroChem is aimed to become a top-ranking electrochemistry journal for primary research papers and critical secondary information from authors across the world. The journal covers the entire scope of pure and applied electrochemistry, the latter encompassing (among others) energy applications, electrochemistry at interfaces (including surfaces), photoelectrochemistry and bioelectrochemistry.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
