Min Cheol Kim, Rachel Gate, David S. Lee, Andrew Tolopko, Andrew Lu, Erin Gordon, Eric Shifrut, Pablo E. Garcia-Nieto, Alexander Marson, Vasilis Ntranos, Chun Jimmie Ye
{"title":"用于单细胞 RNA 测序数据差异表达分析的矩方法框架","authors":"Min Cheol Kim, Rachel Gate, David S. Lee, Andrew Tolopko, Andrew Lu, Erin Gordon, Eric Shifrut, Pablo E. Garcia-Nieto, Alexander Marson, Vasilis Ntranos, Chun Jimmie Ye","doi":"10.1016/j.cell.2024.09.044","DOIUrl":null,"url":null,"abstract":"Differential expression analysis of single-cell RNA sequencing (scRNA-seq) data is central for characterizing how experimental factors affect the distribution of gene expression. However, distinguishing between biological and technical sources of cell-cell variability and assessing the statistical significance of quantitative comparisons between cell groups remain challenging. We introduce Memento, a tool for robust and efficient differential analysis of mean expression, variability, and gene correlation from scRNA-seq data, scalable to millions of cells and thousands of samples. We applied Memento to 70,000 tracheal epithelial cells to identify interferon-responsive genes, 160,000 CRISPR-Cas9 perturbed T cells to reconstruct gene-regulatory networks, 1.2 million peripheral blood mononuclear cells (PBMCs) to map cell-type-specific quantitative trait loci (QTLs), and the 50-million-cell CELLxGENE Discover corpus to compare arbitrary cell groups. In all cases, Memento identified more significant and reproducible differences in mean expression compared with existing methods. It also identified differences in variability and gene correlation that suggest distinct transcriptional regulation mechanisms imparted by perturbations.","PeriodicalId":9656,"journal":{"name":"Cell","volume":"94 1","pages":""},"PeriodicalIF":42.5000,"publicationDate":"2024-10-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Method of moments framework for differential expression analysis of single-cell RNA sequencing data\",\"authors\":\"Min Cheol Kim, Rachel Gate, David S. Lee, Andrew Tolopko, Andrew Lu, Erin Gordon, Eric Shifrut, Pablo E. Garcia-Nieto, Alexander Marson, Vasilis Ntranos, Chun Jimmie Ye\",\"doi\":\"10.1016/j.cell.2024.09.044\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Differential expression analysis of single-cell RNA sequencing (scRNA-seq) data is central for characterizing how experimental factors affect the distribution of gene expression. However, distinguishing between biological and technical sources of cell-cell variability and assessing the statistical significance of quantitative comparisons between cell groups remain challenging. We introduce Memento, a tool for robust and efficient differential analysis of mean expression, variability, and gene correlation from scRNA-seq data, scalable to millions of cells and thousands of samples. We applied Memento to 70,000 tracheal epithelial cells to identify interferon-responsive genes, 160,000 CRISPR-Cas9 perturbed T cells to reconstruct gene-regulatory networks, 1.2 million peripheral blood mononuclear cells (PBMCs) to map cell-type-specific quantitative trait loci (QTLs), and the 50-million-cell CELLxGENE Discover corpus to compare arbitrary cell groups. In all cases, Memento identified more significant and reproducible differences in mean expression compared with existing methods. It also identified differences in variability and gene correlation that suggest distinct transcriptional regulation mechanisms imparted by perturbations.\",\"PeriodicalId\":9656,\"journal\":{\"name\":\"Cell\",\"volume\":\"94 1\",\"pages\":\"\"},\"PeriodicalIF\":42.5000,\"publicationDate\":\"2024-10-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cell\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1016/j.cell.2024.09.044\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.cell.2024.09.044","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Method of moments framework for differential expression analysis of single-cell RNA sequencing data
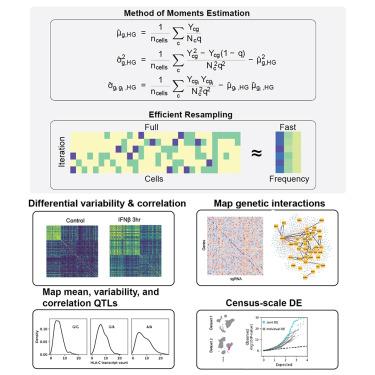
Differential expression analysis of single-cell RNA sequencing (scRNA-seq) data is central for characterizing how experimental factors affect the distribution of gene expression. However, distinguishing between biological and technical sources of cell-cell variability and assessing the statistical significance of quantitative comparisons between cell groups remain challenging. We introduce Memento, a tool for robust and efficient differential analysis of mean expression, variability, and gene correlation from scRNA-seq data, scalable to millions of cells and thousands of samples. We applied Memento to 70,000 tracheal epithelial cells to identify interferon-responsive genes, 160,000 CRISPR-Cas9 perturbed T cells to reconstruct gene-regulatory networks, 1.2 million peripheral blood mononuclear cells (PBMCs) to map cell-type-specific quantitative trait loci (QTLs), and the 50-million-cell CELLxGENE Discover corpus to compare arbitrary cell groups. In all cases, Memento identified more significant and reproducible differences in mean expression compared with existing methods. It also identified differences in variability and gene correlation that suggest distinct transcriptional regulation mechanisms imparted by perturbations.
期刊介绍:
Cells is an international, peer-reviewed, open access journal that focuses on cell biology, molecular biology, and biophysics. It is affiliated with several societies, including the Spanish Society for Biochemistry and Molecular Biology (SEBBM), Nordic Autophagy Society (NAS), Spanish Society of Hematology and Hemotherapy (SEHH), and Society for Regenerative Medicine (Russian Federation) (RPO).
The journal publishes research findings of significant importance in various areas of experimental biology, such as cell biology, molecular biology, neuroscience, immunology, virology, microbiology, cancer, human genetics, systems biology, signaling, and disease mechanisms and therapeutics. The primary criterion for considering papers is whether the results contribute to significant conceptual advances or raise thought-provoking questions and hypotheses related to interesting and important biological inquiries.
In addition to primary research articles presented in four formats, Cells also features review and opinion articles in its "leading edge" section, discussing recent research advancements and topics of interest to its wide readership.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
