{"title":"靶向 S100A9-TLR2 轴控制脂肪肝缺血再灌注损伤中巨噬细胞 NLRP3 炎症小体的激活","authors":"Mingwei Sheng, Weihua Liu, Yingli Cao, Shixuan Wang, Yuanbang Lin, Wenli Yu","doi":"10.1097/SHK.0000000000002470","DOIUrl":null,"url":null,"abstract":"<p><strong>Abstract: </strong>Liver ischemia reperfusion (IR) injury significantly impacts clinical outcomes by increasing the risk of hepatic dysfunction after liver surgery. Fatty livers are more susceptible to IR stress. Recent studies have demonstrated that S100A9 plays a crucial role in both IR injury and the progression of liver steatosis. Nevertheless, the precise mechanisms underlying these effects remain unclear. In our study, transcriptome analysis of fatty livers subjected to IR insult in mice identified S100A9 as an important mediator. Employing loss-of-function approaches, we investigated the immune regulatory function of S100A9 and its downstream signaling in fatty liver IR injury. As expected, S100A9 emerged as one of the most significantly upregulated genes during the reperfusion stage in fatty livers. Genetic knockdown of S100A9 markedly ameliorated liver pathological damage, evidenced by reduced macrophage/neutrophil infiltration as well as the decreased expression of proinflammatory factors. Transcriptome/functional studies revealed that S100A9 triggered liver inflammatory response via regulating toll-like receptor 2 (TLR2)/activating transcription factor 4 (ATF4) signaling. Additionally, TLR2 expression was notably increased in macrophages from ischemic fatty livers. In vitro , recombinant S100A9-stimulated macrophages exhibited the elevated production of proinflammatory factors and TLR2/ATF4 pathway activation. Intriguingly, S100A9 facilitated ATF4 nuclear translocation and enhanced NEK7/NLRP3 inflammasome activation in macrophages. In conclusion, our study identified S100A9 as a key regulator responsible for macrophage NLRP3 inflammasome activation and subsequent inflammatory injury in fatty liver IR process. Targeting TLR2/ATF4 signaling may offer a novel therapeutic strategy for mitigating S100A9-mediated liver injury.</p>","PeriodicalId":21667,"journal":{"name":"SHOCK","volume":" ","pages":"292-298"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11776876/pdf/","citationCount":"0","resultStr":"{\"title\":\"TARGETING S100A9-TLR2 AXIS CONTROLS MACROPHAGE NLRP3 INFLAMMASOME ACTIVATION IN FATTY LIVER ISCHEMIA REPERFUSION INJURY.\",\"authors\":\"Mingwei Sheng, Weihua Liu, Yingli Cao, Shixuan Wang, Yuanbang Lin, Wenli Yu\",\"doi\":\"10.1097/SHK.0000000000002470\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Abstract: </strong>Liver ischemia reperfusion (IR) injury significantly impacts clinical outcomes by increasing the risk of hepatic dysfunction after liver surgery. Fatty livers are more susceptible to IR stress. Recent studies have demonstrated that S100A9 plays a crucial role in both IR injury and the progression of liver steatosis. Nevertheless, the precise mechanisms underlying these effects remain unclear. In our study, transcriptome analysis of fatty livers subjected to IR insult in mice identified S100A9 as an important mediator. Employing loss-of-function approaches, we investigated the immune regulatory function of S100A9 and its downstream signaling in fatty liver IR injury. As expected, S100A9 emerged as one of the most significantly upregulated genes during the reperfusion stage in fatty livers. Genetic knockdown of S100A9 markedly ameliorated liver pathological damage, evidenced by reduced macrophage/neutrophil infiltration as well as the decreased expression of proinflammatory factors. Transcriptome/functional studies revealed that S100A9 triggered liver inflammatory response via regulating toll-like receptor 2 (TLR2)/activating transcription factor 4 (ATF4) signaling. Additionally, TLR2 expression was notably increased in macrophages from ischemic fatty livers. In vitro , recombinant S100A9-stimulated macrophages exhibited the elevated production of proinflammatory factors and TLR2/ATF4 pathway activation. Intriguingly, S100A9 facilitated ATF4 nuclear translocation and enhanced NEK7/NLRP3 inflammasome activation in macrophages. In conclusion, our study identified S100A9 as a key regulator responsible for macrophage NLRP3 inflammasome activation and subsequent inflammatory injury in fatty liver IR process. Targeting TLR2/ATF4 signaling may offer a novel therapeutic strategy for mitigating S100A9-mediated liver injury.</p>\",\"PeriodicalId\":21667,\"journal\":{\"name\":\"SHOCK\",\"volume\":\" \",\"pages\":\"292-298\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11776876/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"SHOCK\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1097/SHK.0000000000002470\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CRITICAL CARE MEDICINE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"SHOCK","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1097/SHK.0000000000002470","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CRITICAL CARE MEDICINE","Score":null,"Total":0}
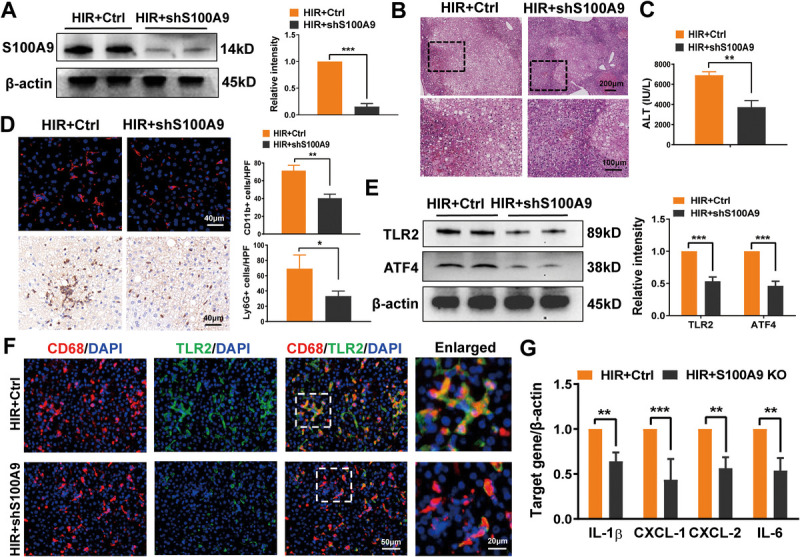
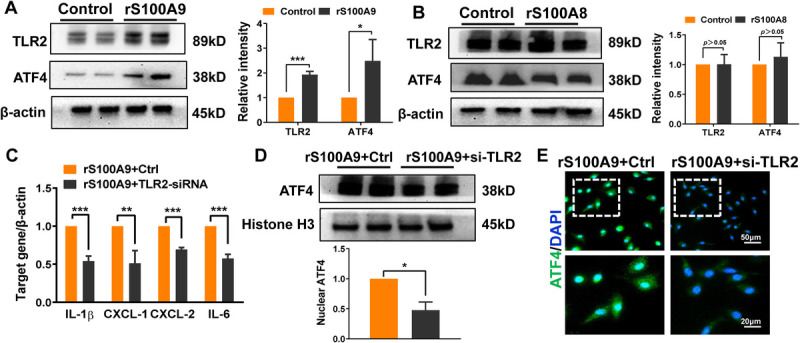
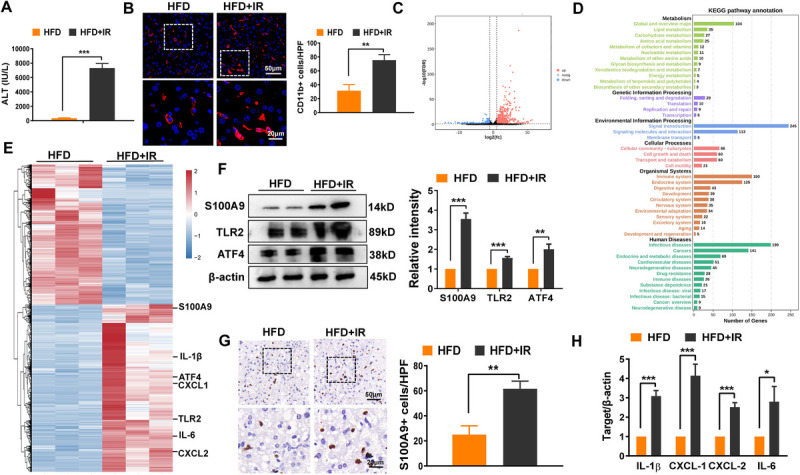
Abstract: Liver ischemia reperfusion (IR) injury significantly impacts clinical outcomes by increasing the risk of hepatic dysfunction after liver surgery. Fatty livers are more susceptible to IR stress. Recent studies have demonstrated that S100A9 plays a crucial role in both IR injury and the progression of liver steatosis. Nevertheless, the precise mechanisms underlying these effects remain unclear. In our study, transcriptome analysis of fatty livers subjected to IR insult in mice identified S100A9 as an important mediator. Employing loss-of-function approaches, we investigated the immune regulatory function of S100A9 and its downstream signaling in fatty liver IR injury. As expected, S100A9 emerged as one of the most significantly upregulated genes during the reperfusion stage in fatty livers. Genetic knockdown of S100A9 markedly ameliorated liver pathological damage, evidenced by reduced macrophage/neutrophil infiltration as well as the decreased expression of proinflammatory factors. Transcriptome/functional studies revealed that S100A9 triggered liver inflammatory response via regulating toll-like receptor 2 (TLR2)/activating transcription factor 4 (ATF4) signaling. Additionally, TLR2 expression was notably increased in macrophages from ischemic fatty livers. In vitro , recombinant S100A9-stimulated macrophages exhibited the elevated production of proinflammatory factors and TLR2/ATF4 pathway activation. Intriguingly, S100A9 facilitated ATF4 nuclear translocation and enhanced NEK7/NLRP3 inflammasome activation in macrophages. In conclusion, our study identified S100A9 as a key regulator responsible for macrophage NLRP3 inflammasome activation and subsequent inflammatory injury in fatty liver IR process. Targeting TLR2/ATF4 signaling may offer a novel therapeutic strategy for mitigating S100A9-mediated liver injury.
期刊介绍:
SHOCK®: Injury, Inflammation, and Sepsis: Laboratory and Clinical Approaches includes studies of novel therapeutic approaches, such as immunomodulation, gene therapy, nutrition, and others. The mission of the Journal is to foster and promote multidisciplinary studies, both experimental and clinical in nature, that critically examine the etiology, mechanisms and novel therapeutics of shock-related pathophysiological conditions. Its purpose is to excel as a vehicle for timely publication in the areas of basic and clinical studies of shock, trauma, sepsis, inflammation, ischemia, and related pathobiological states, with particular emphasis on the biologic mechanisms that determine the response to such injury. Making such information available will ultimately facilitate improved care of the traumatized or septic individual.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
