{"title":"多中心骨溶解结节病(MONA):病例系列和文献综述。","authors":"Mahabaleshwar Mamadapur, Sabarinath Mahadevan, Ponniah Subramanian ArulRajamurugan, Srilakshmi Gandham, Swati Singh","doi":"10.31138/mjr.311203.mon","DOIUrl":null,"url":null,"abstract":"<p><p>Multicentric Osteolysis Nodulosis and Arthropathy (MONA) is a rare skeletal disorder driven by mutations in the MMP2 gene, leading to bone and joint degradation. This case series presents three unique MONA cases, highlighting clinical, radiological, and genetic aspects. These insights shed light on the complexities of MONA, aiding early diagnosis and multidisciplinary management. Case 1 is a 13-year-old male, born to consanguineous parents, presented with a 5-year history of progressive joint deformities, pain, and difficulty walking. Initially diagnosed as juvenile idiopathic arthritis (JIA), despite treatment, his symptoms persisted. Examination revealed multiple clinical findings, including joint contractures and nodules. Genetic analysis identified a pathogenic variant in the MMP2 gene, confirming MONA. Case 2 and Case 3 were two siblings, aged 12 and 17 years respectively, who presented progressive joint contractures in their hands and feet since early childhood. Clinical examinations revealed contractures and subcutaneous nodules. Genetic analysis confirmed MONA with a shared homozygous pathogenic MMP2 variant, emphasising the genetic basis of this rare disorder.</p>","PeriodicalId":32816,"journal":{"name":"Mediterranean Journal of Rheumatology","volume":"35 3","pages":"486-489"},"PeriodicalIF":0.0000,"publicationDate":"2024-07-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11500111/pdf/","citationCount":"0","resultStr":"{\"title\":\"Multicentric Osteolysis Nodulosis and Arthropathy (MONA): A Case Series and Review of the Literature.\",\"authors\":\"Mahabaleshwar Mamadapur, Sabarinath Mahadevan, Ponniah Subramanian ArulRajamurugan, Srilakshmi Gandham, Swati Singh\",\"doi\":\"10.31138/mjr.311203.mon\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Multicentric Osteolysis Nodulosis and Arthropathy (MONA) is a rare skeletal disorder driven by mutations in the MMP2 gene, leading to bone and joint degradation. This case series presents three unique MONA cases, highlighting clinical, radiological, and genetic aspects. These insights shed light on the complexities of MONA, aiding early diagnosis and multidisciplinary management. Case 1 is a 13-year-old male, born to consanguineous parents, presented with a 5-year history of progressive joint deformities, pain, and difficulty walking. Initially diagnosed as juvenile idiopathic arthritis (JIA), despite treatment, his symptoms persisted. Examination revealed multiple clinical findings, including joint contractures and nodules. Genetic analysis identified a pathogenic variant in the MMP2 gene, confirming MONA. Case 2 and Case 3 were two siblings, aged 12 and 17 years respectively, who presented progressive joint contractures in their hands and feet since early childhood. Clinical examinations revealed contractures and subcutaneous nodules. Genetic analysis confirmed MONA with a shared homozygous pathogenic MMP2 variant, emphasising the genetic basis of this rare disorder.</p>\",\"PeriodicalId\":32816,\"journal\":{\"name\":\"Mediterranean Journal of Rheumatology\",\"volume\":\"35 3\",\"pages\":\"486-489\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-07-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11500111/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Mediterranean Journal of Rheumatology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.31138/mjr.311203.mon\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mediterranean Journal of Rheumatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.31138/mjr.311203.mon","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
Multicentric Osteolysis Nodulosis and Arthropathy (MONA): A Case Series and Review of the Literature.
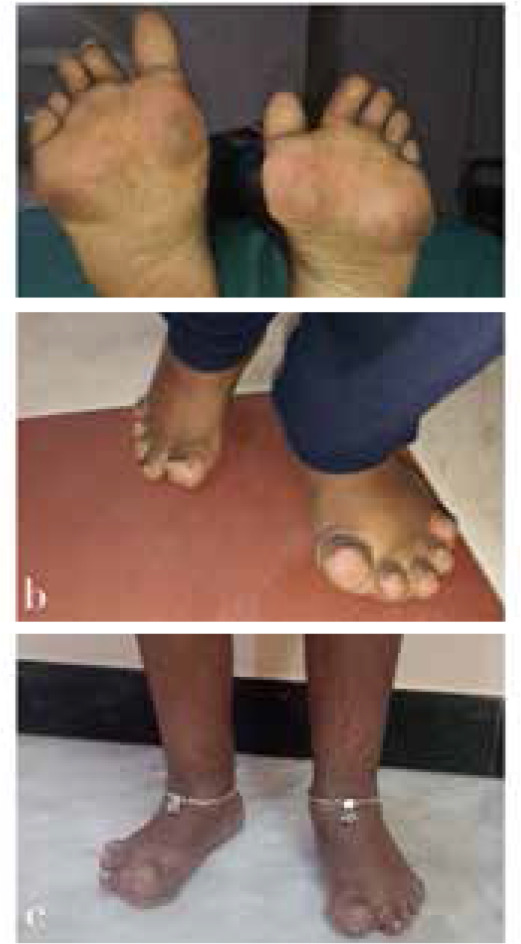
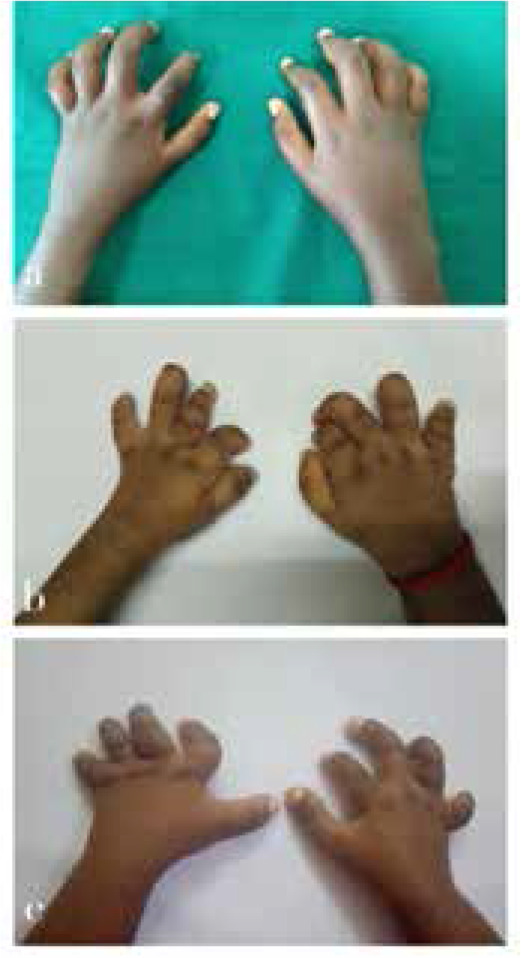
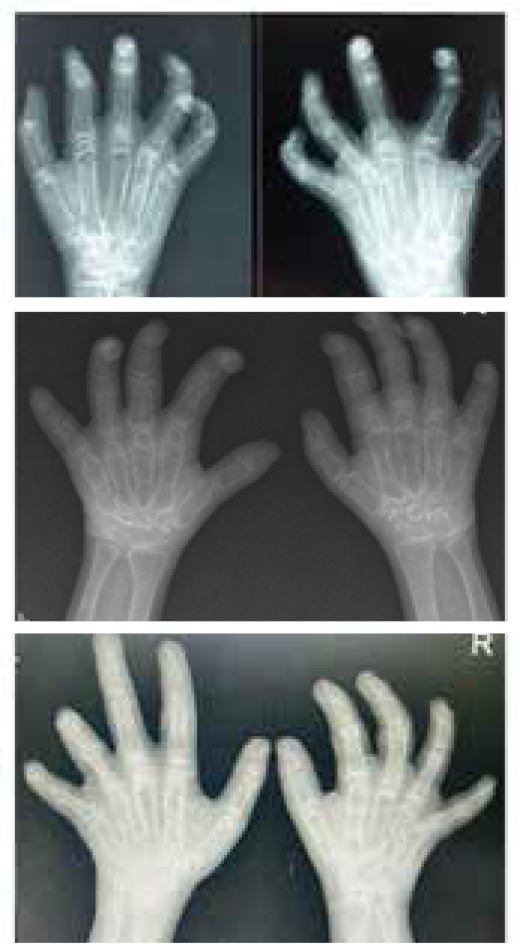
Multicentric Osteolysis Nodulosis and Arthropathy (MONA) is a rare skeletal disorder driven by mutations in the MMP2 gene, leading to bone and joint degradation. This case series presents three unique MONA cases, highlighting clinical, radiological, and genetic aspects. These insights shed light on the complexities of MONA, aiding early diagnosis and multidisciplinary management. Case 1 is a 13-year-old male, born to consanguineous parents, presented with a 5-year history of progressive joint deformities, pain, and difficulty walking. Initially diagnosed as juvenile idiopathic arthritis (JIA), despite treatment, his symptoms persisted. Examination revealed multiple clinical findings, including joint contractures and nodules. Genetic analysis identified a pathogenic variant in the MMP2 gene, confirming MONA. Case 2 and Case 3 were two siblings, aged 12 and 17 years respectively, who presented progressive joint contractures in their hands and feet since early childhood. Clinical examinations revealed contractures and subcutaneous nodules. Genetic analysis confirmed MONA with a shared homozygous pathogenic MMP2 variant, emphasising the genetic basis of this rare disorder.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
