Christina Klötzer, Franziska Schnabel, Anne-Sophie Kubasch, Madlen Jentzsch, Georg-Nikolaus Franke, Jens Uhlig, Helene Faust, Robin-Tobias Jauss, Henry Oppermann, Denny Popp, Klaus H Metzeler, Johannes R Lemke, Vladan Vučinić, Uwe Platzbecker
{"title":"模仿骨髓增生异常肿瘤(MDS)的硫胺素反应性巨幼红细胞贫血(TRMA)综合征。","authors":"Christina Klötzer, Franziska Schnabel, Anne-Sophie Kubasch, Madlen Jentzsch, Georg-Nikolaus Franke, Jens Uhlig, Helene Faust, Robin-Tobias Jauss, Henry Oppermann, Denny Popp, Klaus H Metzeler, Johannes R Lemke, Vladan Vučinić, Uwe Platzbecker","doi":"10.1159/000542286","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Thiamine-responsive megaloblastic anemia syndrome (TRMA) is a rare autosomal recessive disease with a homozygous or compound-heterozygous mutation in the SLC19A2 gene characterized by megaloblastic anemia, diabetes mellitus (DM), and sensorineural hearing loss with onset in childhood. Folic acid and vitamin B12 in serum are normal with dysplastic erythropoiesis in the bone marrow often mimicking myelodysplastic neoplasms (MDS) as a potential differential diagnosis. Thiamine substitution leads to normalization of anemia, without effects on hearing loss or DM.</p><p><strong>Case presentation: </strong>We report about a 38-year-old male patient, presented with a 12-year history of anemia, insulin dependent DM, optic neuropathy, and a cataract since early childhood. The laboratory showed megaloblastic anemia. Other values were normal. The bone marrow smear showed dysplastic erythropoiesis with megaloblastic changes, and normal findings in cytogenetic and molecular genetic examinations. Next-generation sequencing-based diagnostics revealed a heterozygous missense variant in the SLC19A2 gene on the maternal allele and a 3.4 Mb inversion in the chromosomal region 1q24.2 with breaking points in FAM78B and SLC19A2 on the paternal allele. Treatment with oral thiamine 100 mg daily was initiated, and 12 weeks later hemoglobin levels and bone marrow morphology had normalized.</p><p><strong>Conclusion: </strong>Late-onset TRMA should be considered in adult patients with indicative comorbidities and a typical phenotype, which may mimic features of MDS.</p>","PeriodicalId":6981,"journal":{"name":"Acta Haematologica","volume":" ","pages":"380-385"},"PeriodicalIF":1.1000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12306944/pdf/","citationCount":"0","resultStr":"{\"title\":\"Thiamine-Responsive Megaloblastic Anemia Syndrome Mimicking Myelodysplastic Neoplasm.\",\"authors\":\"Christina Klötzer, Franziska Schnabel, Anne-Sophie Kubasch, Madlen Jentzsch, Georg-Nikolaus Franke, Jens Uhlig, Helene Faust, Robin-Tobias Jauss, Henry Oppermann, Denny Popp, Klaus H Metzeler, Johannes R Lemke, Vladan Vučinić, Uwe Platzbecker\",\"doi\":\"10.1159/000542286\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Thiamine-responsive megaloblastic anemia syndrome (TRMA) is a rare autosomal recessive disease with a homozygous or compound-heterozygous mutation in the SLC19A2 gene characterized by megaloblastic anemia, diabetes mellitus (DM), and sensorineural hearing loss with onset in childhood. Folic acid and vitamin B12 in serum are normal with dysplastic erythropoiesis in the bone marrow often mimicking myelodysplastic neoplasms (MDS) as a potential differential diagnosis. Thiamine substitution leads to normalization of anemia, without effects on hearing loss or DM.</p><p><strong>Case presentation: </strong>We report about a 38-year-old male patient, presented with a 12-year history of anemia, insulin dependent DM, optic neuropathy, and a cataract since early childhood. The laboratory showed megaloblastic anemia. Other values were normal. The bone marrow smear showed dysplastic erythropoiesis with megaloblastic changes, and normal findings in cytogenetic and molecular genetic examinations. Next-generation sequencing-based diagnostics revealed a heterozygous missense variant in the SLC19A2 gene on the maternal allele and a 3.4 Mb inversion in the chromosomal region 1q24.2 with breaking points in FAM78B and SLC19A2 on the paternal allele. Treatment with oral thiamine 100 mg daily was initiated, and 12 weeks later hemoglobin levels and bone marrow morphology had normalized.</p><p><strong>Conclusion: </strong>Late-onset TRMA should be considered in adult patients with indicative comorbidities and a typical phenotype, which may mimic features of MDS.</p>\",\"PeriodicalId\":6981,\"journal\":{\"name\":\"Acta Haematologica\",\"volume\":\" \",\"pages\":\"380-385\"},\"PeriodicalIF\":1.1000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12306944/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Haematologica\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1159/000542286\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Haematologica","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1159/000542286","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Introduction: Thiamine-responsive megaloblastic anemia syndrome (TRMA) is a rare autosomal recessive disease with a homozygous or compound-heterozygous mutation in the SLC19A2 gene characterized by megaloblastic anemia, diabetes mellitus (DM), and sensorineural hearing loss with onset in childhood. Folic acid and vitamin B12 in serum are normal with dysplastic erythropoiesis in the bone marrow often mimicking myelodysplastic neoplasms (MDS) as a potential differential diagnosis. Thiamine substitution leads to normalization of anemia, without effects on hearing loss or DM.
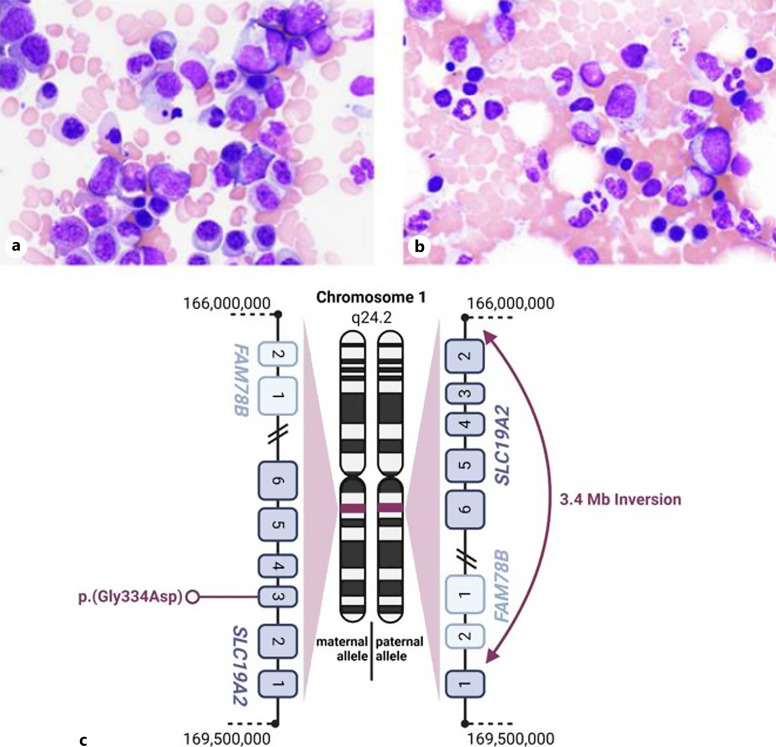
Case presentation: We report about a 38-year-old male patient, presented with a 12-year history of anemia, insulin dependent DM, optic neuropathy, and a cataract since early childhood. The laboratory showed megaloblastic anemia. Other values were normal. The bone marrow smear showed dysplastic erythropoiesis with megaloblastic changes, and normal findings in cytogenetic and molecular genetic examinations. Next-generation sequencing-based diagnostics revealed a heterozygous missense variant in the SLC19A2 gene on the maternal allele and a 3.4 Mb inversion in the chromosomal region 1q24.2 with breaking points in FAM78B and SLC19A2 on the paternal allele. Treatment with oral thiamine 100 mg daily was initiated, and 12 weeks later hemoglobin levels and bone marrow morphology had normalized.
Conclusion: Late-onset TRMA should be considered in adult patients with indicative comorbidities and a typical phenotype, which may mimic features of MDS.
期刊介绍:
''Acta Haematologica'' is a well-established and internationally recognized clinically-oriented journal featuring balanced, wide-ranging coverage of current hematology research. A wealth of information on such problems as anemia, leukemia, lymphoma, multiple myeloma, hereditary disorders, blood coagulation, growth factors, hematopoiesis and differentiation is contained in first-rate basic and clinical papers some of which are accompanied by editorial comments by eminent experts. These are supplemented by short state-of-the-art communications, reviews and correspondence as well as occasional special issues devoted to ‘hot topics’ in hematology. These will keep the practicing hematologist well informed of the new developments in the field.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
