{"title":"模拟 4H-SiC 中锂的吸附、电子转移以及硅-碳-锂化合物的热力学函数","authors":"S. M. Asadov, S. N. Mustafaeva, V. F. Lukichev","doi":"10.1134/S0036024424701607","DOIUrl":null,"url":null,"abstract":"<p>The adsorption, electronic, and thermodynamic properties of the 2 × 2 × 1 and 3 × 3 × 1 supercells of the binary compounds (<span>\\({{{\\text{A}}}_{n}}{{{\\text{B}}}_{m}} = 4{\\text{H}}- {\\text{SiC}},\\)</span> <span>\\({{\\alpha }}{\\kern 1pt} {\\text{ - }}{\\kern 1pt} {\\text{L}}{{{\\text{i}}}_{2}}{{{\\text{C}}}_{2}},\\)</span> <span>\\({\\text{L}}{{{\\text{i}}}_{n}}{\\text{S}}{{{\\text{i}}}_{m}}\\)</span>) of the Si–C–Li system were studied using the density functional theory (DFT). The theoretical capacity of the 4H–SiC hexagonal polytype was found to exceed that of graphite (370 mA h/g) used as an anode material for lithium-ion batteries. The crystalline compounds <span>\\({{{\\text{A}}}_{n}}{{{\\text{B}}}_{m}}\\)</span> have electronic conductivity. The DFT calculations used the exchange-correlation functional within the framework of the generalized gradient approximation (GGA PBE). The parameters of the crystal structure, the adsorption energy of the <span>\\({\\text{L}}{{{\\text{i}}}_{{{\\text{ads}}}}}\\)</span> adatom on the 4H–SiC substrate, the electronic band structure, and the thermodynamic properties of the supercells of <span>\\({{{\\text{A}}}_{n}}{{{\\text{B}}}_{m}}\\)</span> compounds were calculated. The thermodynamically favorable position of <span>\\({\\text{L}}{{{\\text{i}}}_{{{\\text{ads}}}}}\\)</span> and the stable configuration of the 4H–SiC〈Li<sub>ads</sub>〉 supercells were determined. The DFT calculations of the enthalpy of formation of <span>\\({{{\\text{A}}}_{n}}{{{\\text{B}}}_{m}}\\)</span> compounds in the Si–C–Li ternary system were performed. The calculated characteristics of <span>\\({{{\\text{A}}}_{n}}{{{\\text{B}}}_{m}}\\)</span> agree with the experimental data. The equilibrium connodes in the concentration triangle of Si–C–Li were established using the standard thermodynamic potentials of <span>\\({{{\\text{A}}}_{n}}{{{\\text{B}}}_{m}}\\)</span> compounds and the changes in energy in solid-phase exchange reactions between these compounds. An isothermal section of the phase diagram of Si–C–Li at 298 K was constructed.</p>","PeriodicalId":767,"journal":{"name":"Russian Journal of Physical Chemistry A","volume":"98 11","pages":"2437 - 2446"},"PeriodicalIF":0.8000,"publicationDate":"2024-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Simulation of Lithium Adsorption in 4H–SiC, Electron Transfer, and Thermodynamic Functions of Si–C–Li Compounds\",\"authors\":\"S. M. Asadov, S. N. Mustafaeva, V. F. Lukichev\",\"doi\":\"10.1134/S0036024424701607\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The adsorption, electronic, and thermodynamic properties of the 2 × 2 × 1 and 3 × 3 × 1 supercells of the binary compounds (<span>\\\\({{{\\\\text{A}}}_{n}}{{{\\\\text{B}}}_{m}} = 4{\\\\text{H}}- {\\\\text{SiC}},\\\\)</span> <span>\\\\({{\\\\alpha }}{\\\\kern 1pt} {\\\\text{ - }}{\\\\kern 1pt} {\\\\text{L}}{{{\\\\text{i}}}_{2}}{{{\\\\text{C}}}_{2}},\\\\)</span> <span>\\\\({\\\\text{L}}{{{\\\\text{i}}}_{n}}{\\\\text{S}}{{{\\\\text{i}}}_{m}}\\\\)</span>) of the Si–C–Li system were studied using the density functional theory (DFT). The theoretical capacity of the 4H–SiC hexagonal polytype was found to exceed that of graphite (370 mA h/g) used as an anode material for lithium-ion batteries. The crystalline compounds <span>\\\\({{{\\\\text{A}}}_{n}}{{{\\\\text{B}}}_{m}}\\\\)</span> have electronic conductivity. The DFT calculations used the exchange-correlation functional within the framework of the generalized gradient approximation (GGA PBE). The parameters of the crystal structure, the adsorption energy of the <span>\\\\({\\\\text{L}}{{{\\\\text{i}}}_{{{\\\\text{ads}}}}}\\\\)</span> adatom on the 4H–SiC substrate, the electronic band structure, and the thermodynamic properties of the supercells of <span>\\\\({{{\\\\text{A}}}_{n}}{{{\\\\text{B}}}_{m}}\\\\)</span> compounds were calculated. The thermodynamically favorable position of <span>\\\\({\\\\text{L}}{{{\\\\text{i}}}_{{{\\\\text{ads}}}}}\\\\)</span> and the stable configuration of the 4H–SiC〈Li<sub>ads</sub>〉 supercells were determined. The DFT calculations of the enthalpy of formation of <span>\\\\({{{\\\\text{A}}}_{n}}{{{\\\\text{B}}}_{m}}\\\\)</span> compounds in the Si–C–Li ternary system were performed. The calculated characteristics of <span>\\\\({{{\\\\text{A}}}_{n}}{{{\\\\text{B}}}_{m}}\\\\)</span> agree with the experimental data. The equilibrium connodes in the concentration triangle of Si–C–Li were established using the standard thermodynamic potentials of <span>\\\\({{{\\\\text{A}}}_{n}}{{{\\\\text{B}}}_{m}}\\\\)</span> compounds and the changes in energy in solid-phase exchange reactions between these compounds. An isothermal section of the phase diagram of Si–C–Li at 298 K was constructed.</p>\",\"PeriodicalId\":767,\"journal\":{\"name\":\"Russian Journal of Physical Chemistry A\",\"volume\":\"98 11\",\"pages\":\"2437 - 2446\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2024-10-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Russian Journal of Physical Chemistry A\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S0036024424701607\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Russian Journal of Physical Chemistry A","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S0036024424701607","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Simulation of Lithium Adsorption in 4H–SiC, Electron Transfer, and Thermodynamic Functions of Si–C–Li Compounds
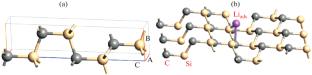
The adsorption, electronic, and thermodynamic properties of the 2 × 2 × 1 and 3 × 3 × 1 supercells of the binary compounds (\({{{\text{A}}}_{n}}{{{\text{B}}}_{m}} = 4{\text{H}}- {\text{SiC}},\)\({{\alpha }}{\kern 1pt} {\text{ - }}{\kern 1pt} {\text{L}}{{{\text{i}}}_{2}}{{{\text{C}}}_{2}},\)\({\text{L}}{{{\text{i}}}_{n}}{\text{S}}{{{\text{i}}}_{m}}\)) of the Si–C–Li system were studied using the density functional theory (DFT). The theoretical capacity of the 4H–SiC hexagonal polytype was found to exceed that of graphite (370 mA h/g) used as an anode material for lithium-ion batteries. The crystalline compounds \({{{\text{A}}}_{n}}{{{\text{B}}}_{m}}\) have electronic conductivity. The DFT calculations used the exchange-correlation functional within the framework of the generalized gradient approximation (GGA PBE). The parameters of the crystal structure, the adsorption energy of the \({\text{L}}{{{\text{i}}}_{{{\text{ads}}}}}\) adatom on the 4H–SiC substrate, the electronic band structure, and the thermodynamic properties of the supercells of \({{{\text{A}}}_{n}}{{{\text{B}}}_{m}}\) compounds were calculated. The thermodynamically favorable position of \({\text{L}}{{{\text{i}}}_{{{\text{ads}}}}}\) and the stable configuration of the 4H–SiC〈Liads〉 supercells were determined. The DFT calculations of the enthalpy of formation of \({{{\text{A}}}_{n}}{{{\text{B}}}_{m}}\) compounds in the Si–C–Li ternary system were performed. The calculated characteristics of \({{{\text{A}}}_{n}}{{{\text{B}}}_{m}}\) agree with the experimental data. The equilibrium connodes in the concentration triangle of Si–C–Li were established using the standard thermodynamic potentials of \({{{\text{A}}}_{n}}{{{\text{B}}}_{m}}\) compounds and the changes in energy in solid-phase exchange reactions between these compounds. An isothermal section of the phase diagram of Si–C–Li at 298 K was constructed.
期刊介绍:
Russian Journal of Physical Chemistry A. Focus on Chemistry (Zhurnal Fizicheskoi Khimii), founded in 1930, offers a comprehensive review of theoretical and experimental research from the Russian Academy of Sciences, leading research and academic centers from Russia and from all over the world.
Articles are devoted to chemical thermodynamics and thermochemistry, biophysical chemistry, photochemistry and magnetochemistry, materials structure, quantum chemistry, physical chemistry of nanomaterials and solutions, surface phenomena and adsorption, and methods and techniques of physicochemical studies.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
