Da-Jiang Liu , Jie Zhang , Long Qi , James W. Evans
{"title":"利用转移学习势对掺氮纳米多孔碳的无序纳米级结构进行分子模拟","authors":"Da-Jiang Liu , Jie Zhang , Long Qi , James W. Evans","doi":"10.1016/j.carbon.2024.119697","DOIUrl":null,"url":null,"abstract":"<div><div>Machine learning (ML)-based molecular dynamics (MD) simulations of the formation of a class of N-doped nanoporous carbons are performed to assess their disordered partially graphitized nanoscale structure. The study is motivated by the effectiveness of so-called nitrogen assembly carbons (NACs) for catalysis applications. Benchmark simulations for pure-C disordered graphitic systems reveal the importance of reliably capturing the vdW component of the potentials in order to accurately describe the tendency for layering of disordered graphene-like sheets. In our modeling, this is achieved by a transfer learning strategy incorporating features of the energetics from the optB88-vdW DFT functional into potentials initially trained with a less expensive functional, thereby providing a superior description of the pure-C systems. Generation from MD simulations of realistic partially graphitized structures is significantly more challenging for N-doped versus for pure C systems. However, such structures are achieved by a tailored MD simulation protocol mimicking the experimental synthesis process and in particular incorporating an annealing and subsequent quenching stages. Simulated PXRD patterns effectively reproduce the features of experimental observations for NACs, including the appearance of a prominent but broad (002) peak at around 25<span><math><msup><mrow></mrow><mrow><mo>∘</mo></mrow></msup></math></span>, and the development of another weaker feature associated with in-layer ordering of mixed C-N graphene-like sheets.</div></div>","PeriodicalId":262,"journal":{"name":"Carbon","volume":"231 ","pages":"Article 119697"},"PeriodicalIF":11.6000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Molecular simulation using transfer-learned potentials for the disordered nanoscale structure of nitrogen-doped nanoporous carbons\",\"authors\":\"Da-Jiang Liu , Jie Zhang , Long Qi , James W. Evans\",\"doi\":\"10.1016/j.carbon.2024.119697\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Machine learning (ML)-based molecular dynamics (MD) simulations of the formation of a class of N-doped nanoporous carbons are performed to assess their disordered partially graphitized nanoscale structure. The study is motivated by the effectiveness of so-called nitrogen assembly carbons (NACs) for catalysis applications. Benchmark simulations for pure-C disordered graphitic systems reveal the importance of reliably capturing the vdW component of the potentials in order to accurately describe the tendency for layering of disordered graphene-like sheets. In our modeling, this is achieved by a transfer learning strategy incorporating features of the energetics from the optB88-vdW DFT functional into potentials initially trained with a less expensive functional, thereby providing a superior description of the pure-C systems. Generation from MD simulations of realistic partially graphitized structures is significantly more challenging for N-doped versus for pure C systems. However, such structures are achieved by a tailored MD simulation protocol mimicking the experimental synthesis process and in particular incorporating an annealing and subsequent quenching stages. Simulated PXRD patterns effectively reproduce the features of experimental observations for NACs, including the appearance of a prominent but broad (002) peak at around 25<span><math><msup><mrow></mrow><mrow><mo>∘</mo></mrow></msup></math></span>, and the development of another weaker feature associated with in-layer ordering of mixed C-N graphene-like sheets.</div></div>\",\"PeriodicalId\":262,\"journal\":{\"name\":\"Carbon\",\"volume\":\"231 \",\"pages\":\"Article 119697\"},\"PeriodicalIF\":11.6000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Carbon\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0008622324009163\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Carbon","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0008622324009163","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/16 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
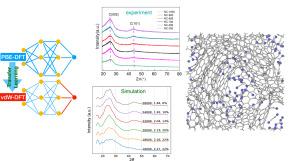
对一类掺氮纳米多孔碳的形成进行了基于机器学习(ML)的分子动力学(MD)模拟,以评估其无序的部分石墨化纳米级结构。这项研究的动机是所谓的氮组装碳(NAC)在催化应用中的有效性。对纯碳无序石墨系统的基准模拟表明,为了准确描述无序石墨烯类薄片的分层趋势,可靠地捕捉电势的 vdW 分量非常重要。在我们的建模中,通过迁移学习策略将 optB88-vdW DFT 函数的能量特征纳入最初用成本较低的函数训练的电位中,从而实现了对纯 C 系统的卓越描述。通过 MD 模拟生成逼真的部分石墨化结构对于掺杂 N 的系统来说比纯 C 系统更具挑战性。不过,通过模仿实验合成过程,特别是结合退火和随后的淬火阶段的定制 MD 模拟协议,这种结构是可以实现的。模拟的 PXRD 图样有效地再现了 NAC 的实验观察特征,包括在 25∘ 左右出现一个突出但宽阔的 (002) 峰,以及与层内混合 C-N 类石墨烯片有序相关的另一个较弱特征。
Molecular simulation using transfer-learned potentials for the disordered nanoscale structure of nitrogen-doped nanoporous carbons
Machine learning (ML)-based molecular dynamics (MD) simulations of the formation of a class of N-doped nanoporous carbons are performed to assess their disordered partially graphitized nanoscale structure. The study is motivated by the effectiveness of so-called nitrogen assembly carbons (NACs) for catalysis applications. Benchmark simulations for pure-C disordered graphitic systems reveal the importance of reliably capturing the vdW component of the potentials in order to accurately describe the tendency for layering of disordered graphene-like sheets. In our modeling, this is achieved by a transfer learning strategy incorporating features of the energetics from the optB88-vdW DFT functional into potentials initially trained with a less expensive functional, thereby providing a superior description of the pure-C systems. Generation from MD simulations of realistic partially graphitized structures is significantly more challenging for N-doped versus for pure C systems. However, such structures are achieved by a tailored MD simulation protocol mimicking the experimental synthesis process and in particular incorporating an annealing and subsequent quenching stages. Simulated PXRD patterns effectively reproduce the features of experimental observations for NACs, including the appearance of a prominent but broad (002) peak at around 25, and the development of another weaker feature associated with in-layer ordering of mixed C-N graphene-like sheets.
期刊介绍:
The journal Carbon is an international multidisciplinary forum for communicating scientific advances in the field of carbon materials. It reports new findings related to the formation, structure, properties, behaviors, and technological applications of carbons. Carbons are a broad class of ordered or disordered solid phases composed primarily of elemental carbon, including but not limited to carbon black, carbon fibers and filaments, carbon nanotubes, diamond and diamond-like carbon, fullerenes, glassy carbon, graphite, graphene, graphene-oxide, porous carbons, pyrolytic carbon, and other sp2 and non-sp2 hybridized carbon systems. Carbon is the companion title to the open access journal Carbon Trends. Relevant application areas for carbon materials include biology and medicine, catalysis, electronic, optoelectronic, spintronic, high-frequency, and photonic devices, energy storage and conversion systems, environmental applications and water treatment, smart materials and systems, and structural and thermal applications.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
