Valeria A. Litvinova , Vladimir B. Tsvetkov , Yulia L. Volodina , Lyubov G. Dezhenkova , Alina A. Markova , Minh Tuan Nguyen , Alexander S. Tikhomirov , Andrey E. Shchekotikhin
{"title":"萘吲哚-2-羧酰胺作为一种亲脂型 Hetarene-Anthraquinones 化合物,对具有 P-gp 抗性的肿瘤细胞有效","authors":"Valeria A. Litvinova , Vladimir B. Tsvetkov , Yulia L. Volodina , Lyubov G. Dezhenkova , Alina A. Markova , Minh Tuan Nguyen , Alexander S. Tikhomirov , Andrey E. Shchekotikhin","doi":"10.1016/j.ejmech.2024.117013","DOIUrl":null,"url":null,"abstract":"<div><div>The acquisition of multidrug resistance (MDR) to chemotherapy is a major obstacle to successful cancer treatment. Aiming to improve the potency of anthraquinone-derived antitumor compounds against MDR cancer cells, we employed a rational design approach to develop new heteroarene-fused anthraquinones. Shifting the carboxamide group in the naphtho[2,3-<em>f</em>]indole scaffold from the 3-position to 2 increased the lipophilicity and P-glycoprotein (P-gp) binding of the derivatives, potentially enhancing their ability to circumvent P-gp-mediated MDR. To validate the computations, we developed a scheme for heterocyclization into esters of naphtho[2,3-<em>f</em>]indole-2-carboxylic acid, based on the 5-<em>endo</em>-dig cyclization of 2-alkynyl-3-amino-1,4-dimethoxyanthraquinone under mild basic conditions using tetra-<em>n</em>-butylammonium fluoride (TBAF). The synthesized naphthoindole-2-carboxamides, particularly compound <strong>1a</strong> bearing (<em>S</em>)-3-aminopyrrolidine in the carboxamide fragment, demonstrated the highest antiproliferative activity. Most importantly, <strong>1a</strong> suppressed the growth of the P-gp-positive K562/4 leukemia tumor cell line (resistance index = 2.4), while its 3-isomer <strong>LCTA-2640</strong> and <strong>Dox</strong> did not (RI = 125 and 140, respectively). Studies of intracellular uptake and distribution showed that <strong>1a</strong>, unlike its 3-substituted isomer, effectively accumulated in resistant tumor cells, confirming the correlation between <em>in silico</em> and experimental data. The lead compound <strong>1a</strong> interacts with DNA duplex and inhibits topoisomerase 1 but does not induce oxidative stress. Treatment with <strong>1a</strong> increases the population of apoptotic cells in both K562 and K562/4 sublines, regardless of the cell cycle phase. Taken together, this work provides an interesting example of how a little modification in chemical structure can lead to striking differences in antitumor properties. In conclusion, we have identified a potent class of compounds that offer distinct advantages in combating resistant tumor cells.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"281 ","pages":"Article 117013"},"PeriodicalIF":5.9000,"publicationDate":"2025-01-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Naphthoindole-2-carboxamides as a lipophilic chemotype of hetarene-anthraquinones potent against P-gp resistant tumor cells\",\"authors\":\"Valeria A. Litvinova , Vladimir B. Tsvetkov , Yulia L. Volodina , Lyubov G. Dezhenkova , Alina A. Markova , Minh Tuan Nguyen , Alexander S. Tikhomirov , Andrey E. Shchekotikhin\",\"doi\":\"10.1016/j.ejmech.2024.117013\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The acquisition of multidrug resistance (MDR) to chemotherapy is a major obstacle to successful cancer treatment. Aiming to improve the potency of anthraquinone-derived antitumor compounds against MDR cancer cells, we employed a rational design approach to develop new heteroarene-fused anthraquinones. Shifting the carboxamide group in the naphtho[2,3-<em>f</em>]indole scaffold from the 3-position to 2 increased the lipophilicity and P-glycoprotein (P-gp) binding of the derivatives, potentially enhancing their ability to circumvent P-gp-mediated MDR. To validate the computations, we developed a scheme for heterocyclization into esters of naphtho[2,3-<em>f</em>]indole-2-carboxylic acid, based on the 5-<em>endo</em>-dig cyclization of 2-alkynyl-3-amino-1,4-dimethoxyanthraquinone under mild basic conditions using tetra-<em>n</em>-butylammonium fluoride (TBAF). The synthesized naphthoindole-2-carboxamides, particularly compound <strong>1a</strong> bearing (<em>S</em>)-3-aminopyrrolidine in the carboxamide fragment, demonstrated the highest antiproliferative activity. Most importantly, <strong>1a</strong> suppressed the growth of the P-gp-positive K562/4 leukemia tumor cell line (resistance index = 2.4), while its 3-isomer <strong>LCTA-2640</strong> and <strong>Dox</strong> did not (RI = 125 and 140, respectively). Studies of intracellular uptake and distribution showed that <strong>1a</strong>, unlike its 3-substituted isomer, effectively accumulated in resistant tumor cells, confirming the correlation between <em>in silico</em> and experimental data. The lead compound <strong>1a</strong> interacts with DNA duplex and inhibits topoisomerase 1 but does not induce oxidative stress. Treatment with <strong>1a</strong> increases the population of apoptotic cells in both K562 and K562/4 sublines, regardless of the cell cycle phase. Taken together, this work provides an interesting example of how a little modification in chemical structure can lead to striking differences in antitumor properties. In conclusion, we have identified a potent class of compounds that offer distinct advantages in combating resistant tumor cells.</div></div>\",\"PeriodicalId\":314,\"journal\":{\"name\":\"European Journal of Medicinal Chemistry\",\"volume\":\"281 \",\"pages\":\"Article 117013\"},\"PeriodicalIF\":5.9000,\"publicationDate\":\"2025-01-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Journal of Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S022352342400895X\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S022352342400895X","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/30 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
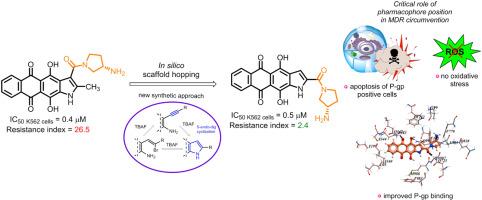
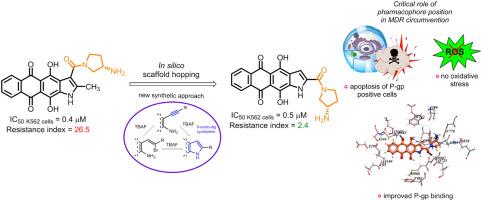
Naphthoindole-2-carboxamides as a lipophilic chemotype of hetarene-anthraquinones potent against P-gp resistant tumor cells
The acquisition of multidrug resistance (MDR) to chemotherapy is a major obstacle to successful cancer treatment. Aiming to improve the potency of anthraquinone-derived antitumor compounds against MDR cancer cells, we employed a rational design approach to develop new heteroarene-fused anthraquinones. Shifting the carboxamide group in the naphtho[2,3-f]indole scaffold from the 3-position to 2 increased the lipophilicity and P-glycoprotein (P-gp) binding of the derivatives, potentially enhancing their ability to circumvent P-gp-mediated MDR. To validate the computations, we developed a scheme for heterocyclization into esters of naphtho[2,3-f]indole-2-carboxylic acid, based on the 5-endo-dig cyclization of 2-alkynyl-3-amino-1,4-dimethoxyanthraquinone under mild basic conditions using tetra-n-butylammonium fluoride (TBAF). The synthesized naphthoindole-2-carboxamides, particularly compound 1a bearing (S)-3-aminopyrrolidine in the carboxamide fragment, demonstrated the highest antiproliferative activity. Most importantly, 1a suppressed the growth of the P-gp-positive K562/4 leukemia tumor cell line (resistance index = 2.4), while its 3-isomer LCTA-2640 and Dox did not (RI = 125 and 140, respectively). Studies of intracellular uptake and distribution showed that 1a, unlike its 3-substituted isomer, effectively accumulated in resistant tumor cells, confirming the correlation between in silico and experimental data. The lead compound 1a interacts with DNA duplex and inhibits topoisomerase 1 but does not induce oxidative stress. Treatment with 1a increases the population of apoptotic cells in both K562 and K562/4 sublines, regardless of the cell cycle phase. Taken together, this work provides an interesting example of how a little modification in chemical structure can lead to striking differences in antitumor properties. In conclusion, we have identified a potent class of compounds that offer distinct advantages in combating resistant tumor cells.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
