{"title":"肾实质马立克氏斑治疗后局灶节段性肾小球硬化的长期观察:病例报告。","authors":"Reiji Takami, Yoshikuni Nagayama, Hiroki Nishiwaki, Toshiharu Ueno, Shigeki Iwasaki, Ashio Yoshimura, Fumihiko Koiwa","doi":"10.1159/000540877","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Malakoplakia is a rare and chronic granulomatous disease that is pathologically characterized by the presence of Michaelis-Gutmann bodies and large macrophage clusters. Malakoplakia of the renal parenchyma is especially rare. In this report, we describe the long-term prognosis of a patient who was diagnosed with and treated for renal parenchymal malakoplakia in infancy.</p><p><strong>Case presentation: </strong>Seventeen years after malakoplakia onset, the patient presented to us with worsening proteinuria. Computed tomography revealed structural abnormalities in the kidney, and focal segmental glomerulosclerosis (FSGS) was diagnosed based on renal biopsy findings. No Michaelis-Gutmann bodies were observed in von Kossa-stained biopsy specimens. Regular outpatient monitoring during the next 9 years showed gradual deterioration of renal function and a moderately high protein/creatinine ratio.</p><p><strong>Conclusion: </strong>Our findings suggest that structural changes due to malakoplakia can cause FSGS. Moreover, structural changes indicate the healing of malakoplakia in infancy and the disappearance of its characteristic lesions over time. Owing to its long-term observation period, this unique case provides new insights into the outcomes of patients with renal parenchymal malakoplakia.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"14 1","pages":"158-163"},"PeriodicalIF":0.9000,"publicationDate":"2024-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11521469/pdf/","citationCount":"0","resultStr":"{\"title\":\"Long-Term Observation of Focal Segmental Glomerulosclerosis after Treatment of Renal Parenchymal Malakoplakia: A Case Report.\",\"authors\":\"Reiji Takami, Yoshikuni Nagayama, Hiroki Nishiwaki, Toshiharu Ueno, Shigeki Iwasaki, Ashio Yoshimura, Fumihiko Koiwa\",\"doi\":\"10.1159/000540877\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Malakoplakia is a rare and chronic granulomatous disease that is pathologically characterized by the presence of Michaelis-Gutmann bodies and large macrophage clusters. Malakoplakia of the renal parenchyma is especially rare. In this report, we describe the long-term prognosis of a patient who was diagnosed with and treated for renal parenchymal malakoplakia in infancy.</p><p><strong>Case presentation: </strong>Seventeen years after malakoplakia onset, the patient presented to us with worsening proteinuria. Computed tomography revealed structural abnormalities in the kidney, and focal segmental glomerulosclerosis (FSGS) was diagnosed based on renal biopsy findings. No Michaelis-Gutmann bodies were observed in von Kossa-stained biopsy specimens. Regular outpatient monitoring during the next 9 years showed gradual deterioration of renal function and a moderately high protein/creatinine ratio.</p><p><strong>Conclusion: </strong>Our findings suggest that structural changes due to malakoplakia can cause FSGS. Moreover, structural changes indicate the healing of malakoplakia in infancy and the disappearance of its characteristic lesions over time. Owing to its long-term observation period, this unique case provides new insights into the outcomes of patients with renal parenchymal malakoplakia.</p>\",\"PeriodicalId\":9599,\"journal\":{\"name\":\"Case Reports in Nephrology and Dialysis\",\"volume\":\"14 1\",\"pages\":\"158-163\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2024-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11521469/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Nephrology and Dialysis\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000540877\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000540877","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
简介马立克氏病是一种罕见的慢性肉芽肿性疾病,其病理特征是存在迈克尔-古特曼体和大型巨噬细胞簇。肾实质的马立克氏病尤其罕见。在本报告中,我们描述了一名在婴儿期被诊断为肾实质恶性肿瘤并接受治疗的患者的长期预后:恶性肿瘤发病 17 年后,患者因蛋白尿加重来我院就诊。计算机断层扫描显示肾脏结构异常,根据肾活检结果诊断为局灶节段性肾小球硬化症(FSGS)。在 von Kossa 染色的活检标本中未发现 Michaelis-Gutmann 体。此后9年的定期门诊监测显示,患者的肾功能逐渐恶化,蛋白/肌酐比值中度偏高:我们的研究结果表明,恶性肿瘤引起的结构变化可导致 FSGS。结论:我们的研究结果表明,恶性肿瘤引起的结构变化可导致 FSGS,而且结构变化表明恶性肿瘤在婴儿期已经愈合,随着时间的推移,其特征性病变会逐渐消失。由于该病例需要长期观察,因此为肾实质恶性肿瘤患者的预后提供了新的视角。
Long-Term Observation of Focal Segmental Glomerulosclerosis after Treatment of Renal Parenchymal Malakoplakia: A Case Report.
Introduction: Malakoplakia is a rare and chronic granulomatous disease that is pathologically characterized by the presence of Michaelis-Gutmann bodies and large macrophage clusters. Malakoplakia of the renal parenchyma is especially rare. In this report, we describe the long-term prognosis of a patient who was diagnosed with and treated for renal parenchymal malakoplakia in infancy.
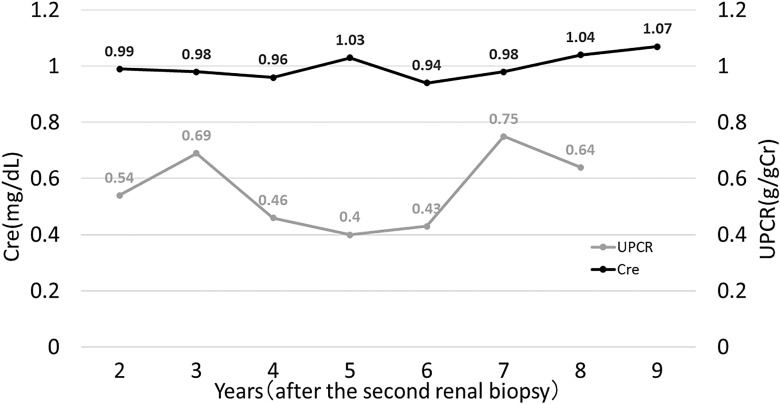
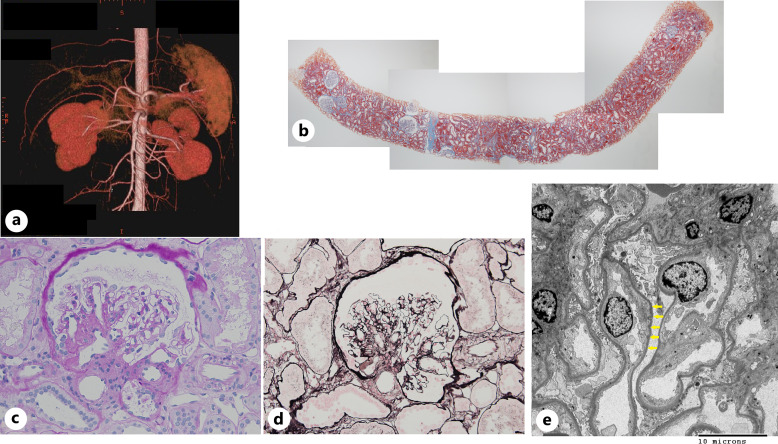
Case presentation: Seventeen years after malakoplakia onset, the patient presented to us with worsening proteinuria. Computed tomography revealed structural abnormalities in the kidney, and focal segmental glomerulosclerosis (FSGS) was diagnosed based on renal biopsy findings. No Michaelis-Gutmann bodies were observed in von Kossa-stained biopsy specimens. Regular outpatient monitoring during the next 9 years showed gradual deterioration of renal function and a moderately high protein/creatinine ratio.
Conclusion: Our findings suggest that structural changes due to malakoplakia can cause FSGS. Moreover, structural changes indicate the healing of malakoplakia in infancy and the disappearance of its characteristic lesions over time. Owing to its long-term observation period, this unique case provides new insights into the outcomes of patients with renal parenchymal malakoplakia.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
