Zongkun Bian, Qiankun Zhang, Haimin Zhang, Fuling Tang, Zhewen Ma, Xuan Lan, Yongchun Luo
{"title":"掺杂 M(M=Al、Pd、Ti、Ge)对非晶硅电子结构和氢扩散行为的影响","authors":"Zongkun Bian, Qiankun Zhang, Haimin Zhang, Fuling Tang, Zhewen Ma, Xuan Lan, Yongchun Luo","doi":"10.1016/j.comptc.2024.114915","DOIUrl":null,"url":null,"abstract":"<div><div>Amorphous silicon (a-Si) models doped with Pd, Ge, Al, and Ti (a-Si/M) at three different doping ratios are generated using <em>Ab initio</em> Molecular Dynamics (AIMD) high-temperature annealing, followed by analysis utilizing first-principles method. The electronic structure calculations reveal strong interactions between Al, Ge and Si, while interaction between Pd, Ti and Si are relatively weak. Moderate doping of Pd and Ti can reconstruct the a-Si surface, reduce the adsorption energy of H on the surface, and increase the Si-Si atomic gap. This will greatly reduce the energy barrier for H to diffuse on the surface and to the subsurface.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114915"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Effect of doping with M (M=Al, Pd, Ti, Ge) on the electronic structure and hydrogen diffusion behavior of amorphous silicon\",\"authors\":\"Zongkun Bian, Qiankun Zhang, Haimin Zhang, Fuling Tang, Zhewen Ma, Xuan Lan, Yongchun Luo\",\"doi\":\"10.1016/j.comptc.2024.114915\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Amorphous silicon (a-Si) models doped with Pd, Ge, Al, and Ti (a-Si/M) at three different doping ratios are generated using <em>Ab initio</em> Molecular Dynamics (AIMD) high-temperature annealing, followed by analysis utilizing first-principles method. The electronic structure calculations reveal strong interactions between Al, Ge and Si, while interaction between Pd, Ti and Si are relatively weak. Moderate doping of Pd and Ti can reconstruct the a-Si surface, reduce the adsorption energy of H on the surface, and increase the Si-Si atomic gap. This will greatly reduce the energy barrier for H to diffuse on the surface and to the subsurface.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114915\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004547\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004547","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/11 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
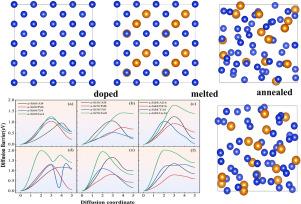
利用 Ab initio 分子动力学(AIMD)高温退火法生成了三种不同掺杂比的掺杂钯、锗、铝和钛(a-Si/M)的非晶硅(a-Si)模型,然后利用第一原理方法进行了分析。电子结构计算显示,Al、Ge 和 Si 之间的相互作用很强,而 Pd、Ti 和 Si 之间的相互作用相对较弱。适度掺杂 Pd 和 Ti 可以重构 a-Si 表面,降低 H 在表面的吸附能,并增大 Si-Si 原子间隙。这将大大降低 H 在表面和向次表面扩散的能量障碍。
Effect of doping with M (M=Al, Pd, Ti, Ge) on the electronic structure and hydrogen diffusion behavior of amorphous silicon
Amorphous silicon (a-Si) models doped with Pd, Ge, Al, and Ti (a-Si/M) at three different doping ratios are generated using Ab initio Molecular Dynamics (AIMD) high-temperature annealing, followed by analysis utilizing first-principles method. The electronic structure calculations reveal strong interactions between Al, Ge and Si, while interaction between Pd, Ti and Si are relatively weak. Moderate doping of Pd and Ti can reconstruct the a-Si surface, reduce the adsorption energy of H on the surface, and increase the Si-Si atomic gap. This will greatly reduce the energy barrier for H to diffuse on the surface and to the subsurface.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
