D. Williamson , S. Ponte , I. Iglesias , N. Tonge , C. Cobas , E.K. Kemsley
{"title":"利用信息传递神经网络 (MPNN) 集合预测 13C NMR 光谱中的化学位移","authors":"D. Williamson , S. Ponte , I. Iglesias , N. Tonge , C. Cobas , E.K. Kemsley","doi":"10.1016/j.jmr.2024.107795","DOIUrl":null,"url":null,"abstract":"<div><div>This study reports a deep learning approach that utilises message passing neural networks (MPNNs) for predicting chemical shifts in <sup>13</sup>C NMR spectra of small molecules. MPNNs were trained on two distinct datasets: one with approximately 4000 labelled structures and another with over 40,000. To reduce stochastic variation, an ensemble framework was implemented, which is simple to deploy on multiple nodes of a High-Performance Computing facility.</div><div>The results emphasise the critical role of training set size and diversity. While prediction performance was comparable on test sets drawn from each dataset, the ensemble trained on the larger dataset retained its accuracy when these sets were crossed over, and when applied to a further collection of approximately 12,000 previously unseen structures introduced after all development work had been completed. In contrast, the ensemble trained on the smaller dataset showed a notable decline in generalisation ability. This difference is attributed to the greater diversity of atomic environments captured in the larger dataset.</div><div>The larger dataset also enabled more robust modelling of various error properties, providing a quantitative foundation for spectral assignment and verification. This was achieved in two ways. First, a clear relationship was observed between prediction errors and the frequency of different node feature vectors in the training data, allowing error estimates to be associated with individual nodes based on their type. These estimates can be used as weights in a modified cityblock distance metric when assigning observed to predicted shifts. Second, the mean absolute prediction error calculated at the structure level is well-fitted by a Gaussian kernel cumulative distribution. This enabled a probabilistic assessment of whether the predicted shifts and assigned observations are consistent with originating from the same molecular structure.</div></div>","PeriodicalId":16267,"journal":{"name":"Journal of magnetic resonance","volume":"368 ","pages":"Article 107795"},"PeriodicalIF":1.9000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Chemical shift prediction in 13C NMR spectroscopy using ensembles of message passing neural networks (MPNNs)\",\"authors\":\"D. Williamson , S. Ponte , I. Iglesias , N. Tonge , C. Cobas , E.K. Kemsley\",\"doi\":\"10.1016/j.jmr.2024.107795\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>This study reports a deep learning approach that utilises message passing neural networks (MPNNs) for predicting chemical shifts in <sup>13</sup>C NMR spectra of small molecules. MPNNs were trained on two distinct datasets: one with approximately 4000 labelled structures and another with over 40,000. To reduce stochastic variation, an ensemble framework was implemented, which is simple to deploy on multiple nodes of a High-Performance Computing facility.</div><div>The results emphasise the critical role of training set size and diversity. While prediction performance was comparable on test sets drawn from each dataset, the ensemble trained on the larger dataset retained its accuracy when these sets were crossed over, and when applied to a further collection of approximately 12,000 previously unseen structures introduced after all development work had been completed. In contrast, the ensemble trained on the smaller dataset showed a notable decline in generalisation ability. This difference is attributed to the greater diversity of atomic environments captured in the larger dataset.</div><div>The larger dataset also enabled more robust modelling of various error properties, providing a quantitative foundation for spectral assignment and verification. This was achieved in two ways. First, a clear relationship was observed between prediction errors and the frequency of different node feature vectors in the training data, allowing error estimates to be associated with individual nodes based on their type. These estimates can be used as weights in a modified cityblock distance metric when assigning observed to predicted shifts. Second, the mean absolute prediction error calculated at the structure level is well-fitted by a Gaussian kernel cumulative distribution. This enabled a probabilistic assessment of whether the predicted shifts and assigned observations are consistent with originating from the same molecular structure.</div></div>\",\"PeriodicalId\":16267,\"journal\":{\"name\":\"Journal of magnetic resonance\",\"volume\":\"368 \",\"pages\":\"Article 107795\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of magnetic resonance\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1090780724001794\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of magnetic resonance","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1090780724001794","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Chemical shift prediction in 13C NMR spectroscopy using ensembles of message passing neural networks (MPNNs)
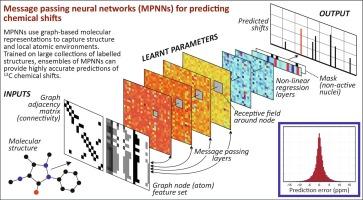
This study reports a deep learning approach that utilises message passing neural networks (MPNNs) for predicting chemical shifts in 13C NMR spectra of small molecules. MPNNs were trained on two distinct datasets: one with approximately 4000 labelled structures and another with over 40,000. To reduce stochastic variation, an ensemble framework was implemented, which is simple to deploy on multiple nodes of a High-Performance Computing facility.
The results emphasise the critical role of training set size and diversity. While prediction performance was comparable on test sets drawn from each dataset, the ensemble trained on the larger dataset retained its accuracy when these sets were crossed over, and when applied to a further collection of approximately 12,000 previously unseen structures introduced after all development work had been completed. In contrast, the ensemble trained on the smaller dataset showed a notable decline in generalisation ability. This difference is attributed to the greater diversity of atomic environments captured in the larger dataset.
The larger dataset also enabled more robust modelling of various error properties, providing a quantitative foundation for spectral assignment and verification. This was achieved in two ways. First, a clear relationship was observed between prediction errors and the frequency of different node feature vectors in the training data, allowing error estimates to be associated with individual nodes based on their type. These estimates can be used as weights in a modified cityblock distance metric when assigning observed to predicted shifts. Second, the mean absolute prediction error calculated at the structure level is well-fitted by a Gaussian kernel cumulative distribution. This enabled a probabilistic assessment of whether the predicted shifts and assigned observations are consistent with originating from the same molecular structure.
期刊介绍:
The Journal of Magnetic Resonance presents original technical and scientific papers in all aspects of magnetic resonance, including nuclear magnetic resonance spectroscopy (NMR) of solids and liquids, electron spin/paramagnetic resonance (EPR), in vivo magnetic resonance imaging (MRI) and spectroscopy (MRS), nuclear quadrupole resonance (NQR) and magnetic resonance phenomena at nearly zero fields or in combination with optics. The Journal''s main aims include deepening the physical principles underlying all these spectroscopies, publishing significant theoretical and experimental results leading to spectral and spatial progress in these areas, and opening new MR-based applications in chemistry, biology and medicine. The Journal also seeks descriptions of novel apparatuses, new experimental protocols, and new procedures of data analysis and interpretation - including computational and quantum-mechanical methods - capable of advancing MR spectroscopy and imaging.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
