Jingjing Lin, Yun-Lu Li, Bo-Li Chen, Hui-Zhen Su, Yi-Heng Zeng, Rui-Huang Zeng, Yu-Duo Zhang, Ru-Kai Chen, Nai-Qing Cai, Yi-Kun Chen, Ru-Ying Yuan, Jun-Yi Jiang, Xiang-Ping Yao, Ning Wang, Wan-Jin Chen, Kang Yang
{"title":"进行性肌阵挛性共济失调是典型的 I 型神经鞘磷脂病(NEU1 基因突变)的最初症状。","authors":"Jingjing Lin, Yun-Lu Li, Bo-Li Chen, Hui-Zhen Su, Yi-Heng Zeng, Rui-Huang Zeng, Yu-Duo Zhang, Ru-Kai Chen, Nai-Qing Cai, Yi-Kun Chen, Ru-Ying Yuan, Jun-Yi Jiang, Xiang-Ping Yao, Ning Wang, Wan-Jin Chen, Kang Yang","doi":"10.1002/acn3.52212","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Expand genetic screening for atypical Type I sialidosis (ST-1) could address its underdiagnosed in both progressive myoclonic ataxia (PMA) and ataxia patients. To evaluate the potential founder effect of mutation in the population.</p><p><strong>Methods: </strong>We enrolled 231 patients with PMA or ataxia from the First Affiliated Hospital of Fujian Medical University. Through Whole Exome Sequencing and Sanger sequencing, we identified the causative gene in patients. Haplotype analysis was employed to explore a potential founder effect of the NEU1 c.544A>G mutation.</p><p><strong>Results: </strong>A total of 31 patients from 23 unrelated families were genetically diagnosed with ST-1. A significant 80.6% of these patients were homozygous for the c.544A>G mutation. We discovered six different NEU1 variants, including two novel mutations: c.951_968del and c.517T>G. The mean age of onset was 18.0 ± 7.1 years. The clinical spectrum of ST-1 featured ataxia and myoclonus as the most common initial symptoms. Over 40% suffered from controlled generalized tonic-clonic seizures. Mobility and independence varied greatly across the cohort. Cherry-red spots were rare, occurring in just 9.5% (2/21) of patients. Brain MRIs were typically unremarkable, except for two patients with unusual findings. EEGs showed diffuse paroxysmal activity in 17 patients. The c.544A>G mutation in NEU1 is a founder variant in Fujian, with a unique haplotype prevalent in East Asians.</p><p><strong>Interpretation: </strong>ST-1 should be suspected in patients with PMA or ataxia in Southeast China, even without macular cherry-red spots and seizures, and the premier test could be a variant screening of the founder variant NEU1 c.544A>G.</p>","PeriodicalId":126,"journal":{"name":"Annals of Clinical and Translational Neurology","volume":" ","pages":"2998-3009"},"PeriodicalIF":3.9000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/acn3.52212","citationCount":"0","resultStr":"{\"title\":\"Progressive myoclonic ataxia as an initial symptom of typical type I sialidosis with NEU1 mutation.\",\"authors\":\"Jingjing Lin, Yun-Lu Li, Bo-Li Chen, Hui-Zhen Su, Yi-Heng Zeng, Rui-Huang Zeng, Yu-Duo Zhang, Ru-Kai Chen, Nai-Qing Cai, Yi-Kun Chen, Ru-Ying Yuan, Jun-Yi Jiang, Xiang-Ping Yao, Ning Wang, Wan-Jin Chen, Kang Yang\",\"doi\":\"10.1002/acn3.52212\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>Expand genetic screening for atypical Type I sialidosis (ST-1) could address its underdiagnosed in both progressive myoclonic ataxia (PMA) and ataxia patients. To evaluate the potential founder effect of mutation in the population.</p><p><strong>Methods: </strong>We enrolled 231 patients with PMA or ataxia from the First Affiliated Hospital of Fujian Medical University. Through Whole Exome Sequencing and Sanger sequencing, we identified the causative gene in patients. Haplotype analysis was employed to explore a potential founder effect of the NEU1 c.544A>G mutation.</p><p><strong>Results: </strong>A total of 31 patients from 23 unrelated families were genetically diagnosed with ST-1. A significant 80.6% of these patients were homozygous for the c.544A>G mutation. We discovered six different NEU1 variants, including two novel mutations: c.951_968del and c.517T>G. The mean age of onset was 18.0 ± 7.1 years. The clinical spectrum of ST-1 featured ataxia and myoclonus as the most common initial symptoms. Over 40% suffered from controlled generalized tonic-clonic seizures. Mobility and independence varied greatly across the cohort. Cherry-red spots were rare, occurring in just 9.5% (2/21) of patients. Brain MRIs were typically unremarkable, except for two patients with unusual findings. EEGs showed diffuse paroxysmal activity in 17 patients. The c.544A>G mutation in NEU1 is a founder variant in Fujian, with a unique haplotype prevalent in East Asians.</p><p><strong>Interpretation: </strong>ST-1 should be suspected in patients with PMA or ataxia in Southeast China, even without macular cherry-red spots and seizures, and the premier test could be a variant screening of the founder variant NEU1 c.544A>G.</p>\",\"PeriodicalId\":126,\"journal\":{\"name\":\"Annals of Clinical and Translational Neurology\",\"volume\":\" \",\"pages\":\"2998-3009\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/acn3.52212\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Clinical and Translational Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/acn3.52212\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Clinical and Translational Neurology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/acn3.52212","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/31 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Progressive myoclonic ataxia as an initial symptom of typical type I sialidosis with NEU1 mutation.
Objective: Expand genetic screening for atypical Type I sialidosis (ST-1) could address its underdiagnosed in both progressive myoclonic ataxia (PMA) and ataxia patients. To evaluate the potential founder effect of mutation in the population.
Methods: We enrolled 231 patients with PMA or ataxia from the First Affiliated Hospital of Fujian Medical University. Through Whole Exome Sequencing and Sanger sequencing, we identified the causative gene in patients. Haplotype analysis was employed to explore a potential founder effect of the NEU1 c.544A>G mutation.
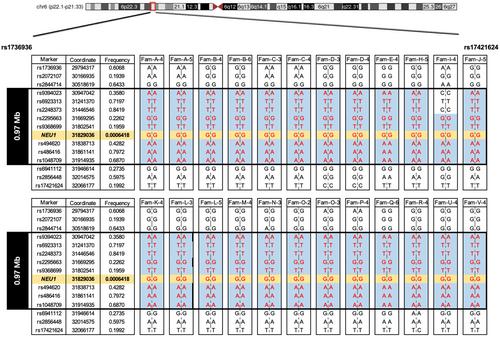
Results: A total of 31 patients from 23 unrelated families were genetically diagnosed with ST-1. A significant 80.6% of these patients were homozygous for the c.544A>G mutation. We discovered six different NEU1 variants, including two novel mutations: c.951_968del and c.517T>G. The mean age of onset was 18.0 ± 7.1 years. The clinical spectrum of ST-1 featured ataxia and myoclonus as the most common initial symptoms. Over 40% suffered from controlled generalized tonic-clonic seizures. Mobility and independence varied greatly across the cohort. Cherry-red spots were rare, occurring in just 9.5% (2/21) of patients. Brain MRIs were typically unremarkable, except for two patients with unusual findings. EEGs showed diffuse paroxysmal activity in 17 patients. The c.544A>G mutation in NEU1 is a founder variant in Fujian, with a unique haplotype prevalent in East Asians.
Interpretation: ST-1 should be suspected in patients with PMA or ataxia in Southeast China, even without macular cherry-red spots and seizures, and the premier test could be a variant screening of the founder variant NEU1 c.544A>G.
期刊介绍:
Annals of Clinical and Translational Neurology is a peer-reviewed journal for rapid dissemination of high-quality research related to all areas of neurology. The journal publishes original research and scholarly reviews focused on the mechanisms and treatments of diseases of the nervous system; high-impact topics in neurologic education; and other topics of interest to the clinical neuroscience community.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
