Jinsei Jung, Sun Young Joo, Hyehyun Min, Jae Won Roh, Kyung Ah Kim, Ji-Hyun Ma, John Hoon Rim, Jung Ah Kim, Se Jin Kim, Seung Hyun Jang, Young Ik Koh, Hye-Youn Kim, Ho Lee, Byoung Choul Kim, Heon Yung Gee, Jinwoong Bok, Jae Young Choi, Je Kyung Seong
{"title":"MYH1 缺乏症会破坏外毛细胞的电流动性,导致听力损失。","authors":"Jinsei Jung, Sun Young Joo, Hyehyun Min, Jae Won Roh, Kyung Ah Kim, Ji-Hyun Ma, John Hoon Rim, Jung Ah Kim, Se Jin Kim, Seung Hyun Jang, Young Ik Koh, Hye-Youn Kim, Ho Lee, Byoung Choul Kim, Heon Yung Gee, Jinwoong Bok, Jae Young Choi, Je Kyung Seong","doi":"10.1038/s12276-024-01338-4","DOIUrl":null,"url":null,"abstract":"Myh1 is a mouse deafness gene with an unknown function in the auditory system. Hearing loss in Myh1-knockout mice is characterized by an elevated threshold for the auditory brainstem response and the absence of a threshold for distortion product otoacoustic emission. Here, we investigated the role of MYH1 in outer hair cells (OHCs), crucial structures in the organ of Corti responsible for regulating cochlear amplification. Direct whole-cell voltage-clamp recordings of OHCs revealed that prestin activity was lower in Myh1-knockout mice than in wild-type mice, indicating abnormal OHC electromotility. We analyzed whole-exome sequencing data from 437 patients with hearing loss of unknown genetic causes and identified biallelic missense variants of MYH1 in five unrelated families. Hearing loss in individuals harboring biallelic MYH1 variants was non-progressive, with an onset ranging from congenital to childhood. Three of five individuals with MYH1 variants displayed osteopenia. Structural prediction by AlphaFold2 followed by molecular dynamic simulations revealed that the identified variants presented structural abnormalities compared with wild-type MYH1. In a heterogeneous overexpression system, MYH1 variants, particularly those in the head domain, abolished MYH1 functions, such as by increasing prestin activity and modulating the membrane traction force. Overall, our findings suggest an essential function of MYH1 in OHCs, as observed in Myh1-deficient mice, and provide genetic evidence linking biallelic MYH1 variants to autosomal recessive hearing loss in humans. Hearing loss is a major health problem affecting many people globally, with a large part due to genes. Researchers studied the MYH1 gene, which was previously associated with hearing in mice but not in humans. The team studied the hearing in mice without the MYH1 gene and looked at genetic data from people with hearing loss. They found mice without the MYH1 gene had hearing problems and impaired hair cell function. They also found several people with changes in the MYH1 gene, providing the first proof of its role in human hearing loss. Research shows how changes in MYH1 affect the function of cells in the inner ear, causing hearing problems in both mice and humans. They hope their work will lead to improved diagnostic tools and treatments for hearing loss. This summary was initially drafted using artificial intelligence, then revised and fact-checked by the author.","PeriodicalId":50466,"journal":{"name":"Experimental and Molecular Medicine","volume":"56 11","pages":"2423-2435"},"PeriodicalIF":12.9000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s12276-024-01338-4.pdf","citationCount":"0","resultStr":"{\"title\":\"MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss\",\"authors\":\"Jinsei Jung, Sun Young Joo, Hyehyun Min, Jae Won Roh, Kyung Ah Kim, Ji-Hyun Ma, John Hoon Rim, Jung Ah Kim, Se Jin Kim, Seung Hyun Jang, Young Ik Koh, Hye-Youn Kim, Ho Lee, Byoung Choul Kim, Heon Yung Gee, Jinwoong Bok, Jae Young Choi, Je Kyung Seong\",\"doi\":\"10.1038/s12276-024-01338-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Myh1 is a mouse deafness gene with an unknown function in the auditory system. Hearing loss in Myh1-knockout mice is characterized by an elevated threshold for the auditory brainstem response and the absence of a threshold for distortion product otoacoustic emission. Here, we investigated the role of MYH1 in outer hair cells (OHCs), crucial structures in the organ of Corti responsible for regulating cochlear amplification. Direct whole-cell voltage-clamp recordings of OHCs revealed that prestin activity was lower in Myh1-knockout mice than in wild-type mice, indicating abnormal OHC electromotility. We analyzed whole-exome sequencing data from 437 patients with hearing loss of unknown genetic causes and identified biallelic missense variants of MYH1 in five unrelated families. Hearing loss in individuals harboring biallelic MYH1 variants was non-progressive, with an onset ranging from congenital to childhood. Three of five individuals with MYH1 variants displayed osteopenia. Structural prediction by AlphaFold2 followed by molecular dynamic simulations revealed that the identified variants presented structural abnormalities compared with wild-type MYH1. In a heterogeneous overexpression system, MYH1 variants, particularly those in the head domain, abolished MYH1 functions, such as by increasing prestin activity and modulating the membrane traction force. Overall, our findings suggest an essential function of MYH1 in OHCs, as observed in Myh1-deficient mice, and provide genetic evidence linking biallelic MYH1 variants to autosomal recessive hearing loss in humans. Hearing loss is a major health problem affecting many people globally, with a large part due to genes. Researchers studied the MYH1 gene, which was previously associated with hearing in mice but not in humans. The team studied the hearing in mice without the MYH1 gene and looked at genetic data from people with hearing loss. They found mice without the MYH1 gene had hearing problems and impaired hair cell function. They also found several people with changes in the MYH1 gene, providing the first proof of its role in human hearing loss. Research shows how changes in MYH1 affect the function of cells in the inner ear, causing hearing problems in both mice and humans. They hope their work will lead to improved diagnostic tools and treatments for hearing loss. This summary was initially drafted using artificial intelligence, then revised and fact-checked by the author.\",\"PeriodicalId\":50466,\"journal\":{\"name\":\"Experimental and Molecular Medicine\",\"volume\":\"56 11\",\"pages\":\"2423-2435\"},\"PeriodicalIF\":12.9000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s12276-024-01338-4.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Experimental and Molecular Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.nature.com/articles/s12276-024-01338-4\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Experimental and Molecular Medicine","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s12276-024-01338-4","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss
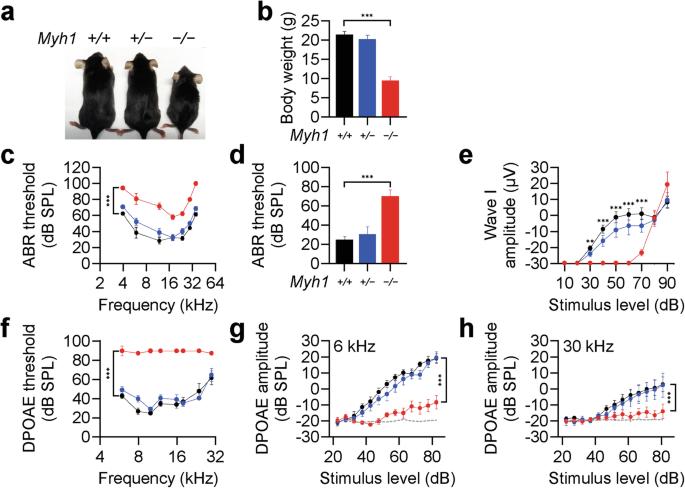
Myh1 is a mouse deafness gene with an unknown function in the auditory system. Hearing loss in Myh1-knockout mice is characterized by an elevated threshold for the auditory brainstem response and the absence of a threshold for distortion product otoacoustic emission. Here, we investigated the role of MYH1 in outer hair cells (OHCs), crucial structures in the organ of Corti responsible for regulating cochlear amplification. Direct whole-cell voltage-clamp recordings of OHCs revealed that prestin activity was lower in Myh1-knockout mice than in wild-type mice, indicating abnormal OHC electromotility. We analyzed whole-exome sequencing data from 437 patients with hearing loss of unknown genetic causes and identified biallelic missense variants of MYH1 in five unrelated families. Hearing loss in individuals harboring biallelic MYH1 variants was non-progressive, with an onset ranging from congenital to childhood. Three of five individuals with MYH1 variants displayed osteopenia. Structural prediction by AlphaFold2 followed by molecular dynamic simulations revealed that the identified variants presented structural abnormalities compared with wild-type MYH1. In a heterogeneous overexpression system, MYH1 variants, particularly those in the head domain, abolished MYH1 functions, such as by increasing prestin activity and modulating the membrane traction force. Overall, our findings suggest an essential function of MYH1 in OHCs, as observed in Myh1-deficient mice, and provide genetic evidence linking biallelic MYH1 variants to autosomal recessive hearing loss in humans. Hearing loss is a major health problem affecting many people globally, with a large part due to genes. Researchers studied the MYH1 gene, which was previously associated with hearing in mice but not in humans. The team studied the hearing in mice without the MYH1 gene and looked at genetic data from people with hearing loss. They found mice without the MYH1 gene had hearing problems and impaired hair cell function. They also found several people with changes in the MYH1 gene, providing the first proof of its role in human hearing loss. Research shows how changes in MYH1 affect the function of cells in the inner ear, causing hearing problems in both mice and humans. They hope their work will lead to improved diagnostic tools and treatments for hearing loss. This summary was initially drafted using artificial intelligence, then revised and fact-checked by the author.
期刊介绍:
Experimental & Molecular Medicine (EMM) stands as Korea's pioneering biochemistry journal, established in 1964 and rejuvenated in 1996 as an Open Access, fully peer-reviewed international journal. Dedicated to advancing translational research and showcasing recent breakthroughs in the biomedical realm, EMM invites submissions encompassing genetic, molecular, and cellular studies of human physiology and diseases. Emphasizing the correlation between experimental and translational research and enhanced clinical benefits, the journal actively encourages contributions employing specific molecular tools. Welcoming studies that bridge basic discoveries with clinical relevance, alongside articles demonstrating clear in vivo significance and novelty, Experimental & Molecular Medicine proudly serves as an open-access, online-only repository of cutting-edge medical research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
