{"title":"系统发育比较分析和蛋白质预测揭示了食源性梭状芽孢杆菌菌株中病毒性因子的分类和多样化分布。","authors":"Ming Zhang, Zhenzhen Yin","doi":"10.1177/11769343241294153","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong><i>Clostridium botulinum</i> and <i>Clostridium perfringens</i>, 2 major foodborne pathogenic fusobacteria, have a variety of virulent protein types with nervous and enterotoxic pathogenic potential, respectively.</p><p><strong>Objective: </strong>The relationship between the molecular evolution of the 2 <i>Clostridium</i> genomes and virulence proteins was studied via a bioinformatics prediction method. The genetic stability, main features of gene coding and structural characteristics of virulence proteins were compared and analyzed to reveal the phylogenetic characteristics, diversity, and distribution of virulence factors of foodborne <i>Clostridium</i> strains.</p><p><strong>Methods: </strong>The phylogenetic analysis was performed via composition vector and average nucleotide identity based methods. Evolutionary distances of virulence genes relative to those of housekeeping genes were calculated via multilocus sequence analysis. Bioinformatics software and tools were used to predict and compare the main functional features of genes encoding virulence proteins, and the structures of virulence proteins were predicted and analyzed through homology modeling and a deep learning algorithm.</p><p><strong>Results: </strong>According to the diversity of toxins, genome evolution tended to cluster based on the protein-coding virulence genes. The evolutionary transfer distances of virulence genes relative to those of housekeeping genes in <i>C. botulinum</i> strains were greater than those in <i>C. perfringens</i> strains, and BoNTs and alpha toxin proteins were located extracellularly. The BoNTs have highly similar structures, but BoNT/A/B and BoNT/E/F have significantly different conformations. The beta2 toxin monomer structure is similar to but simpler than the alpha toxin monomer structure, which has 2 mobile loops in the N-terminal domain. The C-terminal domain of the CPE trimer forms a \"claudin-binding pocket\" shape, which suggests biological relevance, such as in pore formation.</p><p><strong>Conclusions: </strong>According to the genotype of protein-coding virulence genes, the evolution of <i>Clostridium</i> showed a clustering trend. The genetic stability, functional and structural characteristics of foodborne <i>Clostridium</i> virulence proteins reveal the taxonomy and diverse distribution of virulence factors.</p>","PeriodicalId":50472,"journal":{"name":"Evolutionary Bioinformatics","volume":"20 ","pages":"11769343241294153"},"PeriodicalIF":1.5000,"publicationDate":"2024-11-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11536399/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparative Phylogenetic Analysis and Protein Prediction Reveal the Taxonomy and Diverse Distribution of Virulence Factors in Foodborne <i>Clostridium</i> Strains.\",\"authors\":\"Ming Zhang, Zhenzhen Yin\",\"doi\":\"10.1177/11769343241294153\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong><i>Clostridium botulinum</i> and <i>Clostridium perfringens</i>, 2 major foodborne pathogenic fusobacteria, have a variety of virulent protein types with nervous and enterotoxic pathogenic potential, respectively.</p><p><strong>Objective: </strong>The relationship between the molecular evolution of the 2 <i>Clostridium</i> genomes and virulence proteins was studied via a bioinformatics prediction method. The genetic stability, main features of gene coding and structural characteristics of virulence proteins were compared and analyzed to reveal the phylogenetic characteristics, diversity, and distribution of virulence factors of foodborne <i>Clostridium</i> strains.</p><p><strong>Methods: </strong>The phylogenetic analysis was performed via composition vector and average nucleotide identity based methods. Evolutionary distances of virulence genes relative to those of housekeeping genes were calculated via multilocus sequence analysis. Bioinformatics software and tools were used to predict and compare the main functional features of genes encoding virulence proteins, and the structures of virulence proteins were predicted and analyzed through homology modeling and a deep learning algorithm.</p><p><strong>Results: </strong>According to the diversity of toxins, genome evolution tended to cluster based on the protein-coding virulence genes. The evolutionary transfer distances of virulence genes relative to those of housekeeping genes in <i>C. botulinum</i> strains were greater than those in <i>C. perfringens</i> strains, and BoNTs and alpha toxin proteins were located extracellularly. The BoNTs have highly similar structures, but BoNT/A/B and BoNT/E/F have significantly different conformations. The beta2 toxin monomer structure is similar to but simpler than the alpha toxin monomer structure, which has 2 mobile loops in the N-terminal domain. The C-terminal domain of the CPE trimer forms a \\\"claudin-binding pocket\\\" shape, which suggests biological relevance, such as in pore formation.</p><p><strong>Conclusions: </strong>According to the genotype of protein-coding virulence genes, the evolution of <i>Clostridium</i> showed a clustering trend. The genetic stability, functional and structural characteristics of foodborne <i>Clostridium</i> virulence proteins reveal the taxonomy and diverse distribution of virulence factors.</p>\",\"PeriodicalId\":50472,\"journal\":{\"name\":\"Evolutionary Bioinformatics\",\"volume\":\"20 \",\"pages\":\"11769343241294153\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-11-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11536399/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Evolutionary Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1177/11769343241294153\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/11769343241294153","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
Comparative Phylogenetic Analysis and Protein Prediction Reveal the Taxonomy and Diverse Distribution of Virulence Factors in Foodborne Clostridium Strains.
Background: Clostridium botulinum and Clostridium perfringens, 2 major foodborne pathogenic fusobacteria, have a variety of virulent protein types with nervous and enterotoxic pathogenic potential, respectively.
Objective: The relationship between the molecular evolution of the 2 Clostridium genomes and virulence proteins was studied via a bioinformatics prediction method. The genetic stability, main features of gene coding and structural characteristics of virulence proteins were compared and analyzed to reveal the phylogenetic characteristics, diversity, and distribution of virulence factors of foodborne Clostridium strains.
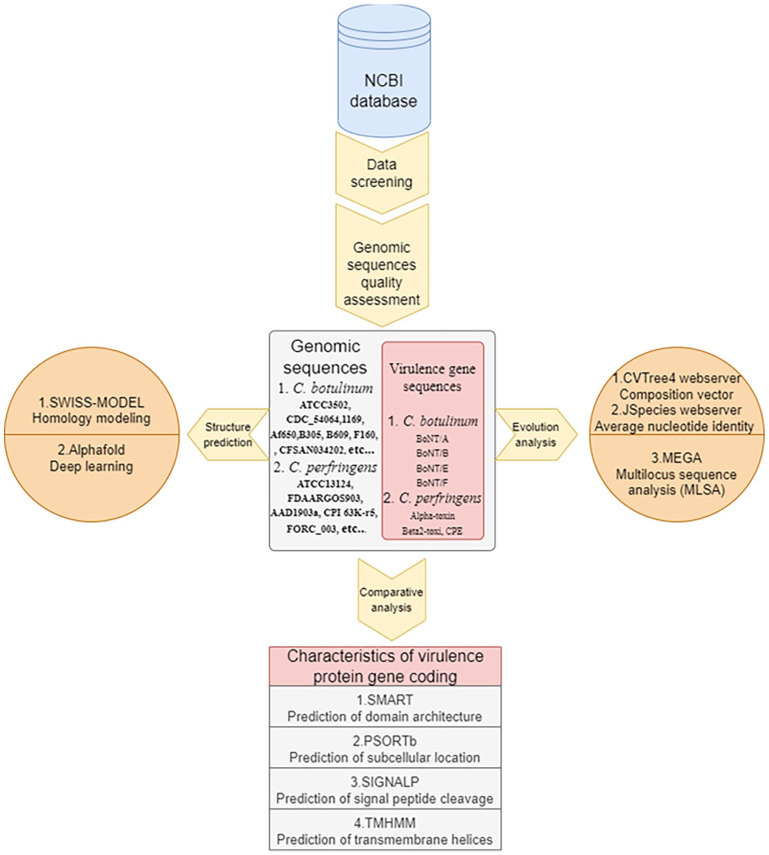
Methods: The phylogenetic analysis was performed via composition vector and average nucleotide identity based methods. Evolutionary distances of virulence genes relative to those of housekeeping genes were calculated via multilocus sequence analysis. Bioinformatics software and tools were used to predict and compare the main functional features of genes encoding virulence proteins, and the structures of virulence proteins were predicted and analyzed through homology modeling and a deep learning algorithm.
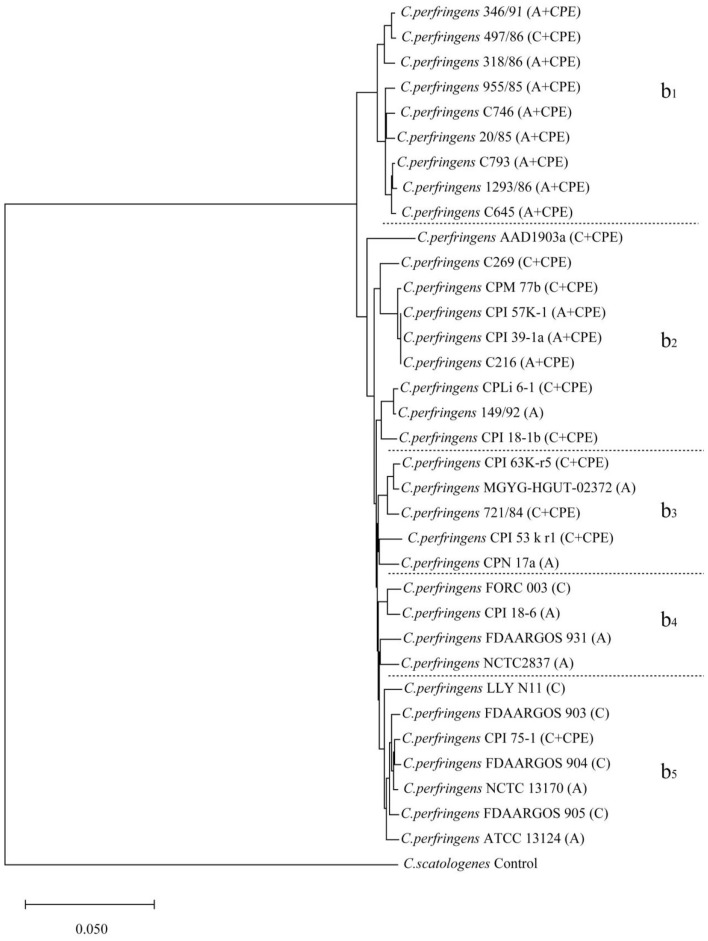
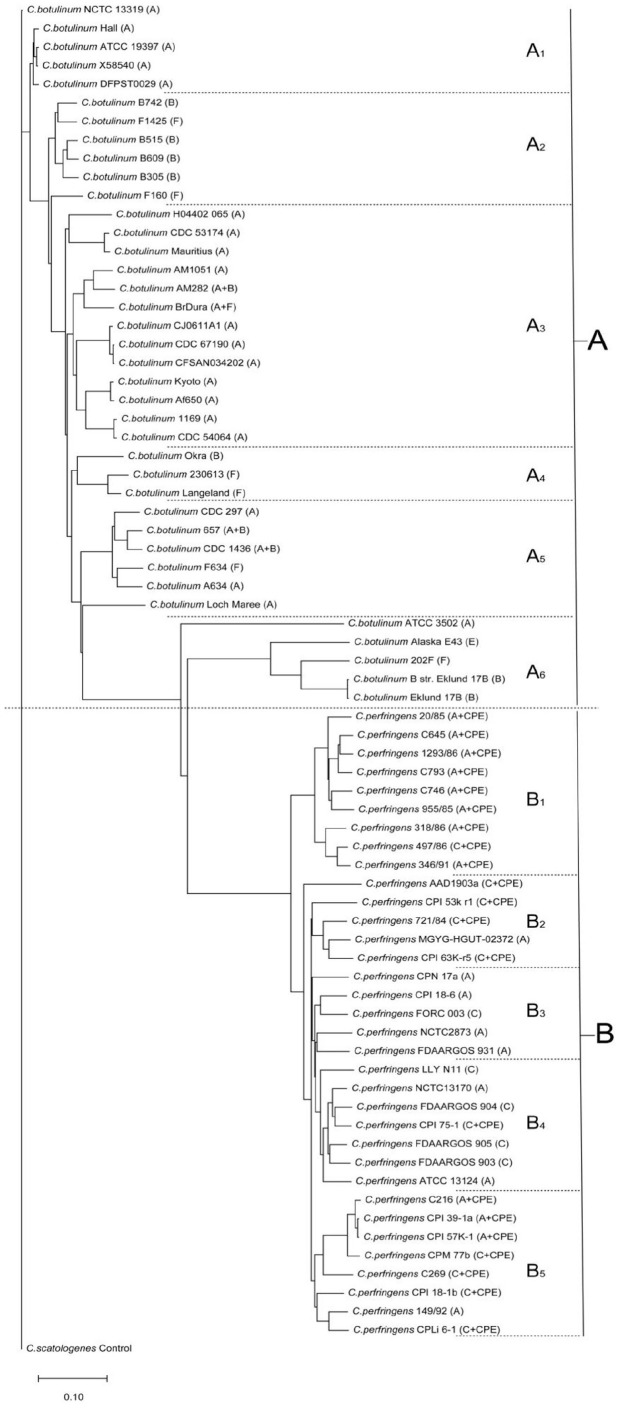
Results: According to the diversity of toxins, genome evolution tended to cluster based on the protein-coding virulence genes. The evolutionary transfer distances of virulence genes relative to those of housekeeping genes in C. botulinum strains were greater than those in C. perfringens strains, and BoNTs and alpha toxin proteins were located extracellularly. The BoNTs have highly similar structures, but BoNT/A/B and BoNT/E/F have significantly different conformations. The beta2 toxin monomer structure is similar to but simpler than the alpha toxin monomer structure, which has 2 mobile loops in the N-terminal domain. The C-terminal domain of the CPE trimer forms a "claudin-binding pocket" shape, which suggests biological relevance, such as in pore formation.
Conclusions: According to the genotype of protein-coding virulence genes, the evolution of Clostridium showed a clustering trend. The genetic stability, functional and structural characteristics of foodborne Clostridium virulence proteins reveal the taxonomy and diverse distribution of virulence factors.
期刊介绍:
Evolutionary Bioinformatics is an open access, peer reviewed international journal focusing on evolutionary bioinformatics. The journal aims to support understanding of organismal form and function through use of molecular, genetic, genomic and proteomic data by giving due consideration to its evolutionary context.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
