{"title":"破解 CHNG3 之谜。","authors":"Satoshi Narumi","doi":"10.6065/apem.2448186.093","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital hypothyroidism (CH), characterized by insufficient thyroid hormone production due to abnormalities in the hypothalamic-pituitary-thyroid axis, is the most common congenital endocrine disorder. We previously conducted comprehensive genetic screening of 102 patients with permanent CH born in Kanagawa Prefecture, Japan and identified mutations in several genes in 19 CH patients, including defects in genes encoding dual oxidase 2, thyroglobulin, thyrotropin receptor, thyroid peroxidase, and paired-box 8. Despite these findings, approximately 80% of cases remain unexplained. CH pedigrees unexplained by known genetic forms of CH have been reported in the literature and registered as congenital hypothyroidism, nongoitrous, 3 (CHNG3; %609893) in Online Mendelian Inheritance in Man. We also identified a Japanese pedigree of CH that was compatible with CHNG3. However, the exact genetic cause of CHNG3 was not revealed by standard analysis methods such as exome sequencing and array comparative genomic hybridization. We therefore took a combined approach and analyzed a total of 11 undiagnosed CH pedigrees by whole genome sequencing to analyze a 3-Mb linkage region, and found a disease-causing variant affecting a TTTG microsatellite in a noncoding region on chromosome 15. Further analysis revealed that 13.9% of 989 Japanese CH patients had abnormalities involving the TTTG microsatellite, with a substantial proportion (41.5%) of familial CH cases carrying these mutations. Identification of the genetic cause of CHNG3 provides new insights into the pathogenesis of CH, and highlights the need for continued exploration of noncoding genomic regions in Mendelian disorders of unknown etiology.</p>","PeriodicalId":44915,"journal":{"name":"Annals of Pediatric Endocrinology & Metabolism","volume":"29 5","pages":"279-283"},"PeriodicalIF":3.3000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11541093/pdf/","citationCount":"0","resultStr":"{\"title\":\"Deciphering the mystery of CHNG3.\",\"authors\":\"Satoshi Narumi\",\"doi\":\"10.6065/apem.2448186.093\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Congenital hypothyroidism (CH), characterized by insufficient thyroid hormone production due to abnormalities in the hypothalamic-pituitary-thyroid axis, is the most common congenital endocrine disorder. We previously conducted comprehensive genetic screening of 102 patients with permanent CH born in Kanagawa Prefecture, Japan and identified mutations in several genes in 19 CH patients, including defects in genes encoding dual oxidase 2, thyroglobulin, thyrotropin receptor, thyroid peroxidase, and paired-box 8. Despite these findings, approximately 80% of cases remain unexplained. CH pedigrees unexplained by known genetic forms of CH have been reported in the literature and registered as congenital hypothyroidism, nongoitrous, 3 (CHNG3; %609893) in Online Mendelian Inheritance in Man. We also identified a Japanese pedigree of CH that was compatible with CHNG3. However, the exact genetic cause of CHNG3 was not revealed by standard analysis methods such as exome sequencing and array comparative genomic hybridization. We therefore took a combined approach and analyzed a total of 11 undiagnosed CH pedigrees by whole genome sequencing to analyze a 3-Mb linkage region, and found a disease-causing variant affecting a TTTG microsatellite in a noncoding region on chromosome 15. Further analysis revealed that 13.9% of 989 Japanese CH patients had abnormalities involving the TTTG microsatellite, with a substantial proportion (41.5%) of familial CH cases carrying these mutations. Identification of the genetic cause of CHNG3 provides new insights into the pathogenesis of CH, and highlights the need for continued exploration of noncoding genomic regions in Mendelian disorders of unknown etiology.</p>\",\"PeriodicalId\":44915,\"journal\":{\"name\":\"Annals of Pediatric Endocrinology & Metabolism\",\"volume\":\"29 5\",\"pages\":\"279-283\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11541093/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Pediatric Endocrinology & Metabolism\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.6065/apem.2448186.093\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Pediatric Endocrinology & Metabolism","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.6065/apem.2448186.093","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/31 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
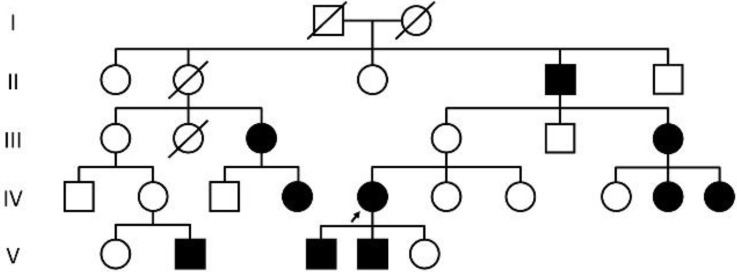
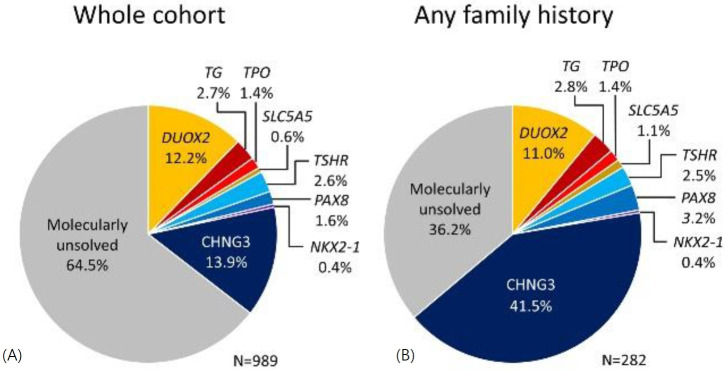
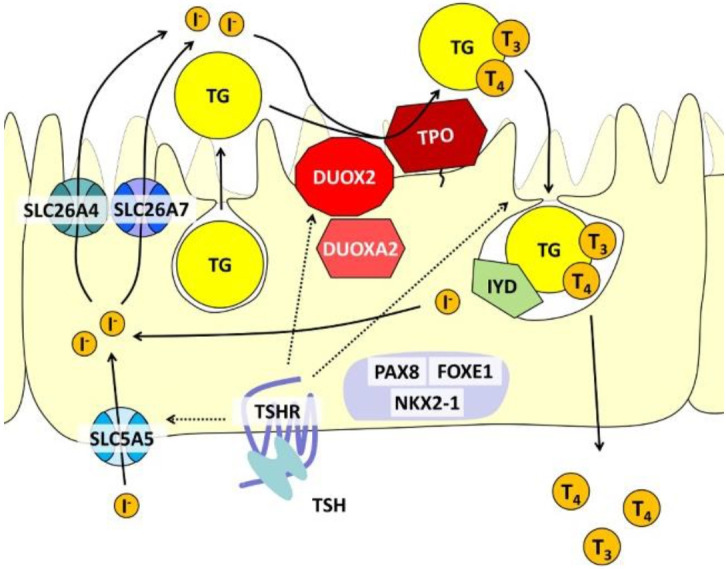
Congenital hypothyroidism (CH), characterized by insufficient thyroid hormone production due to abnormalities in the hypothalamic-pituitary-thyroid axis, is the most common congenital endocrine disorder. We previously conducted comprehensive genetic screening of 102 patients with permanent CH born in Kanagawa Prefecture, Japan and identified mutations in several genes in 19 CH patients, including defects in genes encoding dual oxidase 2, thyroglobulin, thyrotropin receptor, thyroid peroxidase, and paired-box 8. Despite these findings, approximately 80% of cases remain unexplained. CH pedigrees unexplained by known genetic forms of CH have been reported in the literature and registered as congenital hypothyroidism, nongoitrous, 3 (CHNG3; %609893) in Online Mendelian Inheritance in Man. We also identified a Japanese pedigree of CH that was compatible with CHNG3. However, the exact genetic cause of CHNG3 was not revealed by standard analysis methods such as exome sequencing and array comparative genomic hybridization. We therefore took a combined approach and analyzed a total of 11 undiagnosed CH pedigrees by whole genome sequencing to analyze a 3-Mb linkage region, and found a disease-causing variant affecting a TTTG microsatellite in a noncoding region on chromosome 15. Further analysis revealed that 13.9% of 989 Japanese CH patients had abnormalities involving the TTTG microsatellite, with a substantial proportion (41.5%) of familial CH cases carrying these mutations. Identification of the genetic cause of CHNG3 provides new insights into the pathogenesis of CH, and highlights the need for continued exploration of noncoding genomic regions in Mendelian disorders of unknown etiology.
期刊介绍:
The Annals of Pediatric Endocrinology & Metabolism Journal is the official publication of the Korean Society of Pediatric Endocrinology. Its formal abbreviated title is “Ann Pediatr Endocrinol Metab”. It is a peer-reviewed open access journal of medicine published in English. The journal was launched in 1996 under the title of ‘Journal of Korean Society of Pediatric Endocrinology’ until 2011 (pISSN 1226-2242). Since 2012, the title is now changed to ‘Annals of Pediatric Endocrinology & Metabolism’. The Journal is published four times per year on the last day of March, June, September, and December. It is widely distributed for free to members of the Korean Society of Pediatric Endocrinology, medical schools, libraries, and academic institutions. The journal is indexed/tracked/covered by web sites of PubMed Central, PubMed, Emerging Sources Citation Index (ESCI), Scopus, EBSCO, EMBASE, KoreaMed, KoMCI, KCI, Science Central, DOI/CrossRef, Directory of Open Access Journals(DOAJ), and Google Scholar. The aims of Annals of Pediatric Endocrinology & Metabolism are to contribute to the advancements in the fields of pediatric endocrinology & metabolism through the scientific reviews and interchange of all of pediatric endocrinology and metabolism. It aims to reflect the latest clinical, translational, and basic research trends from worldwide valuable achievements. In addition, genome research, epidemiology, public education and clinical practice guidelines in each country are welcomed for publication. The Journal particularly focuses on research conducted with Asian-Pacific children whose genetic and environmental backgrounds are different from those of the Western. Area of specific interest include the following : Growth, puberty, glucose metabolism including diabetes mellitus, obesity, nutrition, disorders of sexual development, pituitary, thyroid, parathyroid, adrenal cortex, bone or other endocrine and metabolic disorders from infancy through adolescence.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
