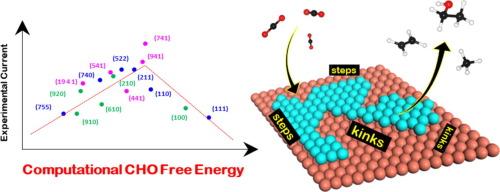
{"title":"铜面二氧化碳还原电化学电流的计算描述符","authors":"Timothy T. Yang, Wissam A. Saidi","doi":"10.1016/j.jcat.2024.115836","DOIUrl":null,"url":null,"abstract":"Computation screening is crucial for designing efficient electrochemical catalysts for carbon dioxide (CO<sub>2</sub>R) reduction that produce valuable hydrocarbons and oxygenates. Herein, leveraging density functional theory calculations of the CO adsorption energy <span><span><math><mrow is=\"true\"><mi is=\"true\" mathvariant=\"normal\">Δ</mi><msub is=\"true\"><mi is=\"true\">E</mi><mrow is=\"true\"><mi is=\"true\" mathvariant=\"normal\">C</mi><mi is=\"true\" mathvariant=\"normal\">O</mi></mrow></msub></mrow></math></span><script type=\"math/mml\"><math><mrow is=\"true\"><mi mathvariant=\"normal\" is=\"true\">Δ</mi><msub is=\"true\"><mi is=\"true\">E</mi><mrow is=\"true\"><mi mathvariant=\"normal\" is=\"true\">C</mi><mi mathvariant=\"normal\" is=\"true\">O</mi></mrow></msub></mrow></math></script></span> on seventeen Cu terminations, we discover a strong linear correlation between <span><span><math><mrow is=\"true\"><mi is=\"true\" mathvariant=\"normal\">Δ</mi><msub is=\"true\"><mi is=\"true\">E</mi><mrow is=\"true\"><mi is=\"true\" mathvariant=\"normal\">C</mi><mi is=\"true\" mathvariant=\"normal\">O</mi></mrow></msub></mrow></math></span><script type=\"math/mml\"><math><mrow is=\"true\"><mi mathvariant=\"normal\" is=\"true\">Δ</mi><msub is=\"true\"><mi is=\"true\">E</mi><mrow is=\"true\"><mi mathvariant=\"normal\" is=\"true\">C</mi><mi mathvariant=\"normal\" is=\"true\">O</mi></mrow></msub></mrow></math></script></span> and the recently experimentally measured CO<sub>2</sub>R electrochemical currents (ACS Catal. 2022, 12, 11, 6578–6588). Examining the <em>ab initio</em> thermodynamics of the early critical intermediates CO*, COH*, and CHO*, we find that CO* <span><span><math><mo is=\"true\" stretchy=\"false\">→</mo></math></span><script type=\"math/mml\"><math><mo stretchy=\"false\" is=\"true\">→</mo></math></script></span> CHO* is the thermodynamically controlling step. <em>Beyond the general CO adsorption energy that only shows a linear trend with CO<sub>2</sub>R activity, we show that the reaction free</em> energy of CO* <span><span><math><mo is=\"true\" stretchy=\"false\">→</mo></math></span><script type=\"math/mml\"><math><mo stretchy=\"false\" is=\"true\">→</mo></math></script></span> CHO* <em>is the descriptor for the overall CO<sub>2</sub>R activity for Cu facets, as it displays a volcano relationship with the experimental current.</em> Importantly, we show that increasing the step and kink density of the Cu terminations not only enhances CO adsorption strength but also modulates the CO* <span><span><math><mo is=\"true\" stretchy=\"false\">→</mo></math></span><script type=\"math/mml\"><math><mo stretchy=\"false\" is=\"true\">→</mo></math></script></span> CHO* pathway, as respectively exemplified in the (941) and (741) facets. In addition, we explain that the high activity of (741) is due to its relatively low hydrogen evolution reaction activity compared with the other Cu surfaces.","PeriodicalId":346,"journal":{"name":"Journal of Catalysis","volume":"35 1","pages":""},"PeriodicalIF":6.5000,"publicationDate":"2024-11-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational descriptor for electrochemical currents of carbon dioxide reduction on Cu facets\",\"authors\":\"Timothy T. Yang, Wissam A. Saidi\",\"doi\":\"10.1016/j.jcat.2024.115836\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Computation screening is crucial for designing efficient electrochemical catalysts for carbon dioxide (CO<sub>2</sub>R) reduction that produce valuable hydrocarbons and oxygenates. Herein, leveraging density functional theory calculations of the CO adsorption energy <span><span><math><mrow is=\\\"true\\\"><mi is=\\\"true\\\" mathvariant=\\\"normal\\\">Δ</mi><msub is=\\\"true\\\"><mi is=\\\"true\\\">E</mi><mrow is=\\\"true\\\"><mi is=\\\"true\\\" mathvariant=\\\"normal\\\">C</mi><mi is=\\\"true\\\" mathvariant=\\\"normal\\\">O</mi></mrow></msub></mrow></math></span><script type=\\\"math/mml\\\"><math><mrow is=\\\"true\\\"><mi mathvariant=\\\"normal\\\" is=\\\"true\\\">Δ</mi><msub is=\\\"true\\\"><mi is=\\\"true\\\">E</mi><mrow is=\\\"true\\\"><mi mathvariant=\\\"normal\\\" is=\\\"true\\\">C</mi><mi mathvariant=\\\"normal\\\" is=\\\"true\\\">O</mi></mrow></msub></mrow></math></script></span> on seventeen Cu terminations, we discover a strong linear correlation between <span><span><math><mrow is=\\\"true\\\"><mi is=\\\"true\\\" mathvariant=\\\"normal\\\">Δ</mi><msub is=\\\"true\\\"><mi is=\\\"true\\\">E</mi><mrow is=\\\"true\\\"><mi is=\\\"true\\\" mathvariant=\\\"normal\\\">C</mi><mi is=\\\"true\\\" mathvariant=\\\"normal\\\">O</mi></mrow></msub></mrow></math></span><script type=\\\"math/mml\\\"><math><mrow is=\\\"true\\\"><mi mathvariant=\\\"normal\\\" is=\\\"true\\\">Δ</mi><msub is=\\\"true\\\"><mi is=\\\"true\\\">E</mi><mrow is=\\\"true\\\"><mi mathvariant=\\\"normal\\\" is=\\\"true\\\">C</mi><mi mathvariant=\\\"normal\\\" is=\\\"true\\\">O</mi></mrow></msub></mrow></math></script></span> and the recently experimentally measured CO<sub>2</sub>R electrochemical currents (ACS Catal. 2022, 12, 11, 6578–6588). Examining the <em>ab initio</em> thermodynamics of the early critical intermediates CO*, COH*, and CHO*, we find that CO* <span><span><math><mo is=\\\"true\\\" stretchy=\\\"false\\\">→</mo></math></span><script type=\\\"math/mml\\\"><math><mo stretchy=\\\"false\\\" is=\\\"true\\\">→</mo></math></script></span> CHO* is the thermodynamically controlling step. <em>Beyond the general CO adsorption energy that only shows a linear trend with CO<sub>2</sub>R activity, we show that the reaction free</em> energy of CO* <span><span><math><mo is=\\\"true\\\" stretchy=\\\"false\\\">→</mo></math></span><script type=\\\"math/mml\\\"><math><mo stretchy=\\\"false\\\" is=\\\"true\\\">→</mo></math></script></span> CHO* <em>is the descriptor for the overall CO<sub>2</sub>R activity for Cu facets, as it displays a volcano relationship with the experimental current.</em> Importantly, we show that increasing the step and kink density of the Cu terminations not only enhances CO adsorption strength but also modulates the CO* <span><span><math><mo is=\\\"true\\\" stretchy=\\\"false\\\">→</mo></math></span><script type=\\\"math/mml\\\"><math><mo stretchy=\\\"false\\\" is=\\\"true\\\">→</mo></math></script></span> CHO* pathway, as respectively exemplified in the (941) and (741) facets. In addition, we explain that the high activity of (741) is due to its relatively low hydrogen evolution reaction activity compared with the other Cu surfaces.\",\"PeriodicalId\":346,\"journal\":{\"name\":\"Journal of Catalysis\",\"volume\":\"35 1\",\"pages\":\"\"},\"PeriodicalIF\":6.5000,\"publicationDate\":\"2024-11-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Catalysis\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1016/j.jcat.2024.115836\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Catalysis","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1016/j.jcat.2024.115836","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
计算筛选对于设计可产生有价值碳氢化合物和含氧化合物的高效二氧化碳(CO2R)还原电化学催化剂至关重要。在此,我们利用密度泛函理论计算了 17 个铜端点上的 CO 吸附能 ΔECOΔECO,发现 ΔECOΔECO 与最近实验测量的 CO2R 电化学电流(ACS Catal.)通过研究早期临界中间产物 CO*、COH* 和 CHO* 的 ab initio 热力学,我们发现 CO* →→ CHO* 是热力学上的控制步骤。除了一般的 CO 吸附能仅与 CO2R 活性呈线性趋势外,我们还发现 CO* →→ CHO* 的反应自由能是铜面整体 CO2R 活性的描述因子,因为它与实验电流呈火山关系。重要的是,我们发现增加 Cu 端面的阶梯和扭结密度不仅能增强 CO 吸附强度,还能调节 CO* →→ CHO* 路径,这在 (941) 和 (741) 面中分别得到了体现。此外,我们还解释了 (741) 面的高活性是由于其氢进化反应活性相对于其他铜表面较低。
Computational descriptor for electrochemical currents of carbon dioxide reduction on Cu facets
Computation screening is crucial for designing efficient electrochemical catalysts for carbon dioxide (CO2R) reduction that produce valuable hydrocarbons and oxygenates. Herein, leveraging density functional theory calculations of the CO adsorption energy on seventeen Cu terminations, we discover a strong linear correlation between and the recently experimentally measured CO2R electrochemical currents (ACS Catal. 2022, 12, 11, 6578–6588). Examining the ab initio thermodynamics of the early critical intermediates CO*, COH*, and CHO*, we find that CO* CHO* is the thermodynamically controlling step. Beyond the general CO adsorption energy that only shows a linear trend with CO2R activity, we show that the reaction free energy of CO* CHO* is the descriptor for the overall CO2R activity for Cu facets, as it displays a volcano relationship with the experimental current. Importantly, we show that increasing the step and kink density of the Cu terminations not only enhances CO adsorption strength but also modulates the CO* CHO* pathway, as respectively exemplified in the (941) and (741) facets. In addition, we explain that the high activity of (741) is due to its relatively low hydrogen evolution reaction activity compared with the other Cu surfaces.
期刊介绍:
The Journal of Catalysis publishes scholarly articles on both heterogeneous and homogeneous catalysis, covering a wide range of chemical transformations. These include various types of catalysis, such as those mediated by photons, plasmons, and electrons. The focus of the studies is to understand the relationship between catalytic function and the underlying chemical properties of surfaces and metal complexes.
The articles in the journal offer innovative concepts and explore the synthesis and kinetics of inorganic solids and homogeneous complexes. Furthermore, they discuss spectroscopic techniques for characterizing catalysts, investigate the interaction of probes and reacting species with catalysts, and employ theoretical methods.
The research presented in the journal should have direct relevance to the field of catalytic processes, addressing either fundamental aspects or applications of catalysis.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
