{"title":"传统流动池设计中 VOSO4 水溶液的结构和动力学:分子动力学模拟研究","authors":"Anwesa Karmakar","doi":"10.1039/D4CP02934H","DOIUrl":null,"url":null,"abstract":"<p >A theoretical model has been proposed to study the structure and dynamics of aqueous vanadyl sulfate (VOSO<small><sub>4</sub></small>) solution used in the conventional flow (CF) through cell design operating under varying thermodynamic conditions. Classical molecular dynamics simulations have been carried out for aqueous solutions of vanadyl sulfate (VOSO<small><sub>4</sub></small>) and sulfuric acid (H<small><sub>2</sub></small>SO<small><sub>4</sub></small>) at two different concentrations and temperatures considering the temperature dependent degree of dissociation of sulfuric acid. The MD trajectories are used to study the equilibrium structural, dynamical properties such as viscosity, diffusivity and surface tension of the aqueous solution of vanadyl sulfate (VOSO<small><sub>4</sub></small>). According to the new model, the cation–cation and cation–anion interaction should be low in order to have a good current density in the conventional flow through cell design and further explains the importance of considering mass transport when designing high energy density redox flow batteries. The model is further validated by calculating the viscosity of each system, individual diffusion coefficient of each ion and by comparing them with the experimental data wherever they are available.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 45","pages":" 28361-28377"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp02934h?page=search","citationCount":"0","resultStr":"{\"title\":\"Structure and dynamics of aqueous VOSO4 solutions in conventional flow through cell design: a molecular dynamics simulation study†\",\"authors\":\"Anwesa Karmakar\",\"doi\":\"10.1039/D4CP02934H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >A theoretical model has been proposed to study the structure and dynamics of aqueous vanadyl sulfate (VOSO<small><sub>4</sub></small>) solution used in the conventional flow (CF) through cell design operating under varying thermodynamic conditions. Classical molecular dynamics simulations have been carried out for aqueous solutions of vanadyl sulfate (VOSO<small><sub>4</sub></small>) and sulfuric acid (H<small><sub>2</sub></small>SO<small><sub>4</sub></small>) at two different concentrations and temperatures considering the temperature dependent degree of dissociation of sulfuric acid. The MD trajectories are used to study the equilibrium structural, dynamical properties such as viscosity, diffusivity and surface tension of the aqueous solution of vanadyl sulfate (VOSO<small><sub>4</sub></small>). According to the new model, the cation–cation and cation–anion interaction should be low in order to have a good current density in the conventional flow through cell design and further explains the importance of considering mass transport when designing high energy density redox flow batteries. The model is further validated by calculating the viscosity of each system, individual diffusion coefficient of each ion and by comparing them with the experimental data wherever they are available.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 45\",\"pages\":\" 28361-28377\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-11-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp02934h?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp02934h\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp02934h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Structure and dynamics of aqueous VOSO4 solutions in conventional flow through cell design: a molecular dynamics simulation study†
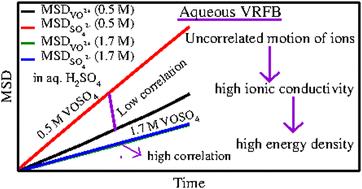
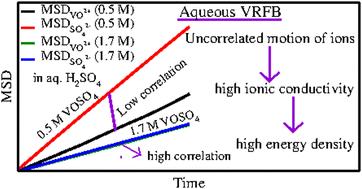
A theoretical model has been proposed to study the structure and dynamics of aqueous vanadyl sulfate (VOSO4) solution used in the conventional flow (CF) through cell design operating under varying thermodynamic conditions. Classical molecular dynamics simulations have been carried out for aqueous solutions of vanadyl sulfate (VOSO4) and sulfuric acid (H2SO4) at two different concentrations and temperatures considering the temperature dependent degree of dissociation of sulfuric acid. The MD trajectories are used to study the equilibrium structural, dynamical properties such as viscosity, diffusivity and surface tension of the aqueous solution of vanadyl sulfate (VOSO4). According to the new model, the cation–cation and cation–anion interaction should be low in order to have a good current density in the conventional flow through cell design and further explains the importance of considering mass transport when designing high energy density redox flow batteries. The model is further validated by calculating the viscosity of each system, individual diffusion coefficient of each ion and by comparing them with the experimental data wherever they are available.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
