Luca Brugnoli, Maxime Ducamp, François-Xavier Coudert
{"title":"基于神经网络的硅质沸石热和机械特性原子间位势研究","authors":"Luca Brugnoli, Maxime Ducamp, François-Xavier Coudert","doi":"10.1021/acs.jpcc.4c07365","DOIUrl":null,"url":null,"abstract":"Molecular dynamics simulations of zeolites are commonly employed for the characterization of their framework dynamics and response to the application of temperature and pressure. While classical interatomic potentials are commonly used for this task, they offer a description of the interactions in the system with limited accuracy. Density functional theory, meanwhile, is accurate, but its high computational expense limits its scalability for large systems or long dynamics. Recent advances in machine learning interatomic potentials, trained on computational data obtained at the quantum chemical level, offer a promising alternative combining high accuracy with computational efficiency. In this study, we developed an MLIP specifically for pure silica zeolites, trained on data from high-temperature ab initio MD simulations across various zeolitic topologies. This MLIP was then applied to predict structural properties, thermal expansion, and pressure response of different zeolites, demonstrating its potential for accuracy and generalizability in simulations of topologies beyond its initial training set.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"98 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Neural Network-Based Interatomic Potential for the Study of Thermal and Mechanical Properties of Siliceous Zeolites\",\"authors\":\"Luca Brugnoli, Maxime Ducamp, François-Xavier Coudert\",\"doi\":\"10.1021/acs.jpcc.4c07365\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Molecular dynamics simulations of zeolites are commonly employed for the characterization of their framework dynamics and response to the application of temperature and pressure. While classical interatomic potentials are commonly used for this task, they offer a description of the interactions in the system with limited accuracy. Density functional theory, meanwhile, is accurate, but its high computational expense limits its scalability for large systems or long dynamics. Recent advances in machine learning interatomic potentials, trained on computational data obtained at the quantum chemical level, offer a promising alternative combining high accuracy with computational efficiency. In this study, we developed an MLIP specifically for pure silica zeolites, trained on data from high-temperature ab initio MD simulations across various zeolitic topologies. This MLIP was then applied to predict structural properties, thermal expansion, and pressure response of different zeolites, demonstrating its potential for accuracy and generalizability in simulations of topologies beyond its initial training set.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"98 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-11-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.4c07365\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c07365","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
沸石的分子动力学模拟通常用于表征其框架动力学以及对温度和压力的响应。虽然经典原子间位势常用于这项任务,但它们对系统中相互作用的描述精度有限。与此同时,密度泛函理论虽然精确,但其高昂的计算费用限制了其在大型系统或长动态过程中的可扩展性。机器学习原子间势的最新进展是在量子化学层面获得的计算数据基础上进行训练,它提供了一种兼具高精度和计算效率的有前途的替代方法。在本研究中,我们开发了一种专门针对纯硅沸石的 MLIP,该 MLIP 是根据各种沸石拓扑结构的高温 ab initio MD 模拟数据进行训练的。然后,我们将该 MLIP 应用于预测不同沸石的结构特性、热膨胀和压力响应,证明了它在模拟其初始训练集之外的拓扑结构时的准确性和通用性潜力。
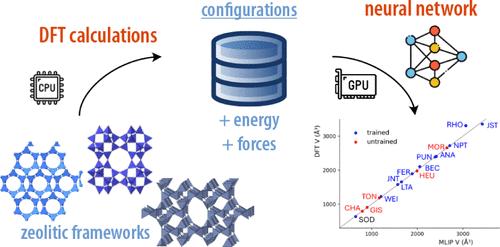
Neural Network-Based Interatomic Potential for the Study of Thermal and Mechanical Properties of Siliceous Zeolites
Molecular dynamics simulations of zeolites are commonly employed for the characterization of their framework dynamics and response to the application of temperature and pressure. While classical interatomic potentials are commonly used for this task, they offer a description of the interactions in the system with limited accuracy. Density functional theory, meanwhile, is accurate, but its high computational expense limits its scalability for large systems or long dynamics. Recent advances in machine learning interatomic potentials, trained on computational data obtained at the quantum chemical level, offer a promising alternative combining high accuracy with computational efficiency. In this study, we developed an MLIP specifically for pure silica zeolites, trained on data from high-temperature ab initio MD simulations across various zeolitic topologies. This MLIP was then applied to predict structural properties, thermal expansion, and pressure response of different zeolites, demonstrating its potential for accuracy and generalizability in simulations of topologies beyond its initial training set.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
