Huiming Xia, My H Hoang, Evelyn Schmidt, Susanna Kiwala, Joshua McMichael, Zachary L Skidmore, Bryan Fisk, Jonathan J Song, Jasreet Hundal, Thomas Mooney, Jason R Walker, S Peter Goedegebuure, Christopher A Miller, William E Gillanders, Obi L Griffith, Malachi Griffith
{"title":"pVACview:高效新抗原优先排序和选择的交互式可视化工具。","authors":"Huiming Xia, My H Hoang, Evelyn Schmidt, Susanna Kiwala, Joshua McMichael, Zachary L Skidmore, Bryan Fisk, Jonathan J Song, Jasreet Hundal, Thomas Mooney, Jason R Walker, S Peter Goedegebuure, Christopher A Miller, William E Gillanders, Obi L Griffith, Malachi Griffith","doi":"10.1186/s13073-024-01384-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Neoantigen-targeting therapies including personalized vaccines have shown promise in the treatment of cancers, particularly when used in combination with checkpoint blockade therapy. At least 100 clinical trials involving these therapies have been initiated globally. Accurate identification and prioritization of neoantigens is crucial for designing these trials, predicting treatment response, and understanding mechanisms of resistance. With the advent of massively parallel DNA and RNA sequencing technologies, it is now possible to computationally predict neoantigens based on patient-specific variant information. However, numerous factors must be considered when prioritizing neoantigens for use in personalized therapies. Complexities such as alternative transcript annotations, various binding, presentation and immunogenicity prediction algorithms, and variable peptide lengths/registers all potentially impact the neoantigen selection process. There has been a rapid development of computational tools that attempt to account for these complexities. While these tools generate numerous algorithmic predictions for neoantigen characterization, results from these pipelines are difficult to navigate and require extensive knowledge of the underlying tools for accurate interpretation. This often leads to over-simplification of pipeline outputs to make them tractable, for example, limiting prediction to a single RNA isoform or only summarizing the top ranked of many possible peptide candidates. In addition to variant detection, gene expression, and predicted peptide binding affinities, recent studies have also demonstrated the importance of mutation location, allele-specific anchor locations, and variation of T-cell response to long versus short peptides. Due to the intricate nature and number of salient neoantigen features, presenting all relevant information to facilitate candidate selection for downstream applications is a difficult challenge that current tools fail to address.</p><p><strong>Results: </strong>We have created pVACview, the first interactive tool designed to aid in the prioritization and selection of neoantigen candidates for personalized neoantigen therapies including cancer vaccines. pVACview has a user-friendly and intuitive interface where users can upload, explore, select, and export their neoantigen candidates. The tool allows users to visualize candidates at multiple levels of detail including variant, transcript, peptide, and algorithm prediction information.</p><p><strong>Conclusions: </strong>pVACview will allow researchers to analyze and prioritize neoantigen candidates with greater efficiency and accuracy in basic and translational settings. The application is available as part of the pVACtools software at pvactools.org and as an online server at pvacview.org.</p>","PeriodicalId":12645,"journal":{"name":"Genome Medicine","volume":"16 1","pages":"132"},"PeriodicalIF":10.4000,"publicationDate":"2024-11-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11562694/pdf/","citationCount":"0","resultStr":"{\"title\":\"pVACview: an interactive visualization tool for efficient neoantigen prioritization and selection.\",\"authors\":\"Huiming Xia, My H Hoang, Evelyn Schmidt, Susanna Kiwala, Joshua McMichael, Zachary L Skidmore, Bryan Fisk, Jonathan J Song, Jasreet Hundal, Thomas Mooney, Jason R Walker, S Peter Goedegebuure, Christopher A Miller, William E Gillanders, Obi L Griffith, Malachi Griffith\",\"doi\":\"10.1186/s13073-024-01384-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Neoantigen-targeting therapies including personalized vaccines have shown promise in the treatment of cancers, particularly when used in combination with checkpoint blockade therapy. At least 100 clinical trials involving these therapies have been initiated globally. Accurate identification and prioritization of neoantigens is crucial for designing these trials, predicting treatment response, and understanding mechanisms of resistance. With the advent of massively parallel DNA and RNA sequencing technologies, it is now possible to computationally predict neoantigens based on patient-specific variant information. However, numerous factors must be considered when prioritizing neoantigens for use in personalized therapies. Complexities such as alternative transcript annotations, various binding, presentation and immunogenicity prediction algorithms, and variable peptide lengths/registers all potentially impact the neoantigen selection process. There has been a rapid development of computational tools that attempt to account for these complexities. While these tools generate numerous algorithmic predictions for neoantigen characterization, results from these pipelines are difficult to navigate and require extensive knowledge of the underlying tools for accurate interpretation. This often leads to over-simplification of pipeline outputs to make them tractable, for example, limiting prediction to a single RNA isoform or only summarizing the top ranked of many possible peptide candidates. In addition to variant detection, gene expression, and predicted peptide binding affinities, recent studies have also demonstrated the importance of mutation location, allele-specific anchor locations, and variation of T-cell response to long versus short peptides. Due to the intricate nature and number of salient neoantigen features, presenting all relevant information to facilitate candidate selection for downstream applications is a difficult challenge that current tools fail to address.</p><p><strong>Results: </strong>We have created pVACview, the first interactive tool designed to aid in the prioritization and selection of neoantigen candidates for personalized neoantigen therapies including cancer vaccines. pVACview has a user-friendly and intuitive interface where users can upload, explore, select, and export their neoantigen candidates. The tool allows users to visualize candidates at multiple levels of detail including variant, transcript, peptide, and algorithm prediction information.</p><p><strong>Conclusions: </strong>pVACview will allow researchers to analyze and prioritize neoantigen candidates with greater efficiency and accuracy in basic and translational settings. The application is available as part of the pVACtools software at pvactools.org and as an online server at pvacview.org.</p>\",\"PeriodicalId\":12645,\"journal\":{\"name\":\"Genome Medicine\",\"volume\":\"16 1\",\"pages\":\"132\"},\"PeriodicalIF\":10.4000,\"publicationDate\":\"2024-11-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11562694/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Medicine\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13073-024-01384-7\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Medicine","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13073-024-01384-7","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:包括个性化疫苗在内的新抗原靶向疗法在治疗癌症方面大有可为,尤其是与检查点阻断疗法联合使用时。全球已启动至少 100 项涉及这些疗法的临床试验。新抗原的准确鉴定和优先排序对于设计这些试验、预测治疗反应和了解抗药性机制至关重要。随着大规模并行 DNA 和 RNA 测序技术的出现,现在可以根据患者特异性变异信息计算预测新抗原。然而,在确定用于个性化疗法的新抗原的优先级时,必须考虑众多因素。替代转录本注释、各种结合、表达和免疫原性预测算法以及可变肽长度/注册等复杂因素都可能影响新抗原的选择过程。试图解释这些复杂性的计算工具发展迅速。虽然这些工具为新抗原特征描述生成了大量算法预测结果,但这些流水线得出的结果却难以驾驭,需要大量的基础工具知识才能准确解读。这往往会导致管道输出结果过于简化,例如,将预测局限于单一 RNA 同工酶,或仅总结许多可能的候选肽中排名最靠前的肽。除了变异检测、基因表达和预测的多肽结合亲和力外,最近的研究还证明了突变位置、等位基因特异性锚定位置以及 T 细胞对长肽和短肽反应的变化的重要性。由于新抗原的特性错综复杂,数量众多,要呈现所有相关信息以方便下游应用的候选选择是一项艰巨的挑战,目前的工具无法解决这一问题:我们创建了 pVACview,这是首个互动式工具,旨在帮助为包括癌症疫苗在内的个性化新抗原疗法确定新抗原候选物的优先级并进行筛选。pVACview 具有用户友好的直观界面,用户可以在此上传、探索、选择和导出其新抗原候选物。结论:pVACview 将使研究人员能够在基础和转化环境中更高效、更准确地分析和优先处理候选新抗原。该应用程序可作为 pVACtools 软件的一部分在 pvactools.org 上下载,也可作为在线服务器在 pvacview.org 上下载。
pVACview: an interactive visualization tool for efficient neoantigen prioritization and selection.
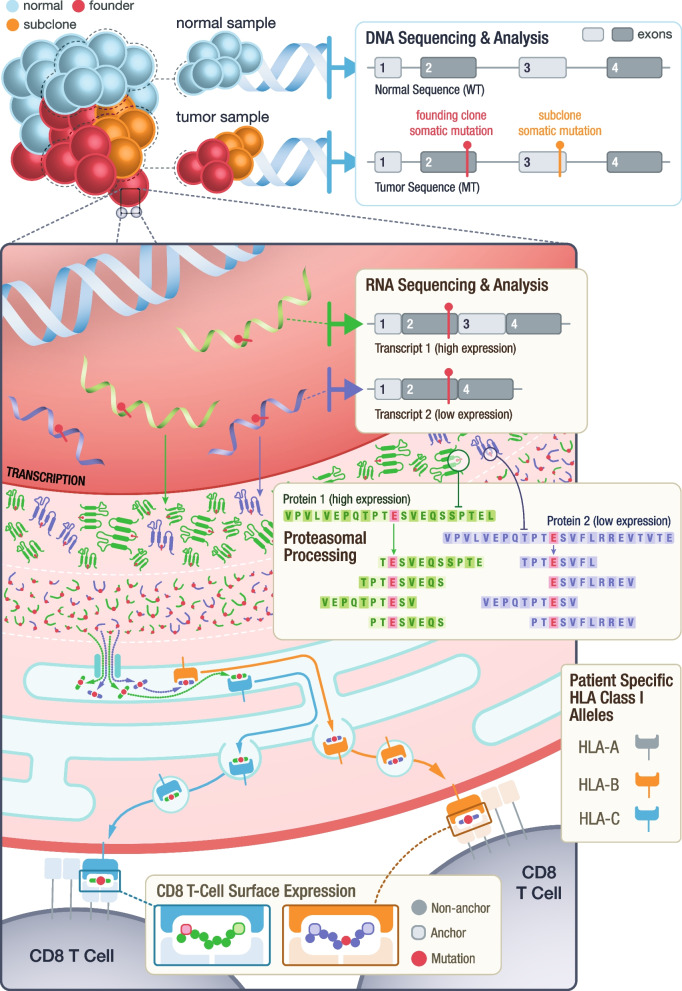
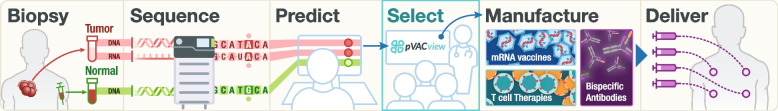
Background: Neoantigen-targeting therapies including personalized vaccines have shown promise in the treatment of cancers, particularly when used in combination with checkpoint blockade therapy. At least 100 clinical trials involving these therapies have been initiated globally. Accurate identification and prioritization of neoantigens is crucial for designing these trials, predicting treatment response, and understanding mechanisms of resistance. With the advent of massively parallel DNA and RNA sequencing technologies, it is now possible to computationally predict neoantigens based on patient-specific variant information. However, numerous factors must be considered when prioritizing neoantigens for use in personalized therapies. Complexities such as alternative transcript annotations, various binding, presentation and immunogenicity prediction algorithms, and variable peptide lengths/registers all potentially impact the neoantigen selection process. There has been a rapid development of computational tools that attempt to account for these complexities. While these tools generate numerous algorithmic predictions for neoantigen characterization, results from these pipelines are difficult to navigate and require extensive knowledge of the underlying tools for accurate interpretation. This often leads to over-simplification of pipeline outputs to make them tractable, for example, limiting prediction to a single RNA isoform or only summarizing the top ranked of many possible peptide candidates. In addition to variant detection, gene expression, and predicted peptide binding affinities, recent studies have also demonstrated the importance of mutation location, allele-specific anchor locations, and variation of T-cell response to long versus short peptides. Due to the intricate nature and number of salient neoantigen features, presenting all relevant information to facilitate candidate selection for downstream applications is a difficult challenge that current tools fail to address.
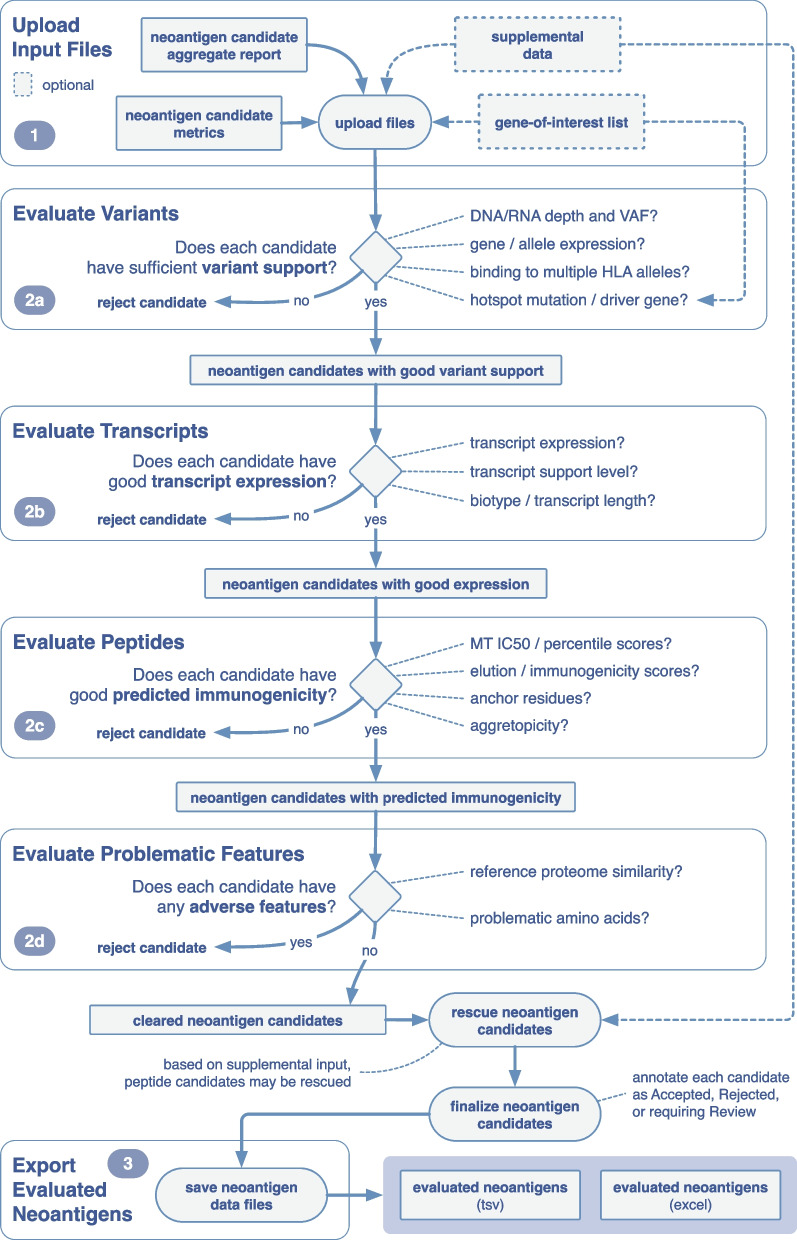
Results: We have created pVACview, the first interactive tool designed to aid in the prioritization and selection of neoantigen candidates for personalized neoantigen therapies including cancer vaccines. pVACview has a user-friendly and intuitive interface where users can upload, explore, select, and export their neoantigen candidates. The tool allows users to visualize candidates at multiple levels of detail including variant, transcript, peptide, and algorithm prediction information.
Conclusions: pVACview will allow researchers to analyze and prioritize neoantigen candidates with greater efficiency and accuracy in basic and translational settings. The application is available as part of the pVACtools software at pvactools.org and as an online server at pvacview.org.
期刊介绍:
Genome Medicine is an open access journal that publishes outstanding research applying genetics, genomics, and multi-omics to understand, diagnose, and treat disease. Bridging basic science and clinical research, it covers areas such as cancer genomics, immuno-oncology, immunogenomics, infectious disease, microbiome, neurogenomics, systems medicine, clinical genomics, gene therapies, precision medicine, and clinical trials. The journal publishes original research, methods, software, and reviews to serve authors and promote broad interest and importance in the field.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
