{"title":"潜在抗病毒药物的计算研究和抗菌活性预测","authors":"Vaithilingam Sasikala , Vadivelu Balachandran , Natarajan Elangovan , Natarajan Arumugam , Abdulrahman I. Almansour","doi":"10.1016/j.molstruc.2024.140711","DOIUrl":null,"url":null,"abstract":"<div><div>The ethyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (S1) was chosen for this investigation due to its important biological and pharmacological activities. Detailed infrared and Raman spectra have been investigated in both theoretical and experimental. Frontier molecular orbitals and the electronic properties of the S1 are explained. The titled compound S1 calculated HOMO-LUMO energy gap is 4.60 eV In molecular electrostatic potential (MEP) map the NO<sub>2</sub> group exhibits a blue color, indicating the presence of nucleophilic sites. The density of state (DOS), non-covalent interaction (NCI), electron localized function (ELF), localized orbital locator (LOL), Mulliken atomic charges, and reactive sites have been investigated. The anti-bacterial, antifungal, antitoxin, antiviral, and antimycobacterial studies have been investigated. The molecular docking investigations of the compound were also conducted. Using the Auto-dock program, the compound S1 molecular docking study has been conducted. The molecular docking study's lowest binding energy is -7.91, -5.62, -6.92, and -5.85 kcal/mol for 4XGK, 4ATO, 3U9 G, and 2DP4 respectively.</div></div>","PeriodicalId":16414,"journal":{"name":"Journal of Molecular Structure","volume":"1323 ","pages":"Article 140711"},"PeriodicalIF":4.7000,"publicationDate":"2025-02-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational investigation and antimicrobial activity prediction of potential antiviral drug\",\"authors\":\"Vaithilingam Sasikala , Vadivelu Balachandran , Natarajan Elangovan , Natarajan Arumugam , Abdulrahman I. Almansour\",\"doi\":\"10.1016/j.molstruc.2024.140711\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The ethyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (S1) was chosen for this investigation due to its important biological and pharmacological activities. Detailed infrared and Raman spectra have been investigated in both theoretical and experimental. Frontier molecular orbitals and the electronic properties of the S1 are explained. The titled compound S1 calculated HOMO-LUMO energy gap is 4.60 eV In molecular electrostatic potential (MEP) map the NO<sub>2</sub> group exhibits a blue color, indicating the presence of nucleophilic sites. The density of state (DOS), non-covalent interaction (NCI), electron localized function (ELF), localized orbital locator (LOL), Mulliken atomic charges, and reactive sites have been investigated. The anti-bacterial, antifungal, antitoxin, antiviral, and antimycobacterial studies have been investigated. The molecular docking investigations of the compound were also conducted. Using the Auto-dock program, the compound S1 molecular docking study has been conducted. The molecular docking study's lowest binding energy is -7.91, -5.62, -6.92, and -5.85 kcal/mol for 4XGK, 4ATO, 3U9 G, and 2DP4 respectively.</div></div>\",\"PeriodicalId\":16414,\"journal\":{\"name\":\"Journal of Molecular Structure\",\"volume\":\"1323 \",\"pages\":\"Article 140711\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2025-02-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Structure\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022286024032198\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/10 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Structure","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022286024032198","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/10 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Computational investigation and antimicrobial activity prediction of potential antiviral drug
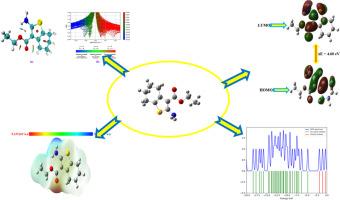
The ethyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (S1) was chosen for this investigation due to its important biological and pharmacological activities. Detailed infrared and Raman spectra have been investigated in both theoretical and experimental. Frontier molecular orbitals and the electronic properties of the S1 are explained. The titled compound S1 calculated HOMO-LUMO energy gap is 4.60 eV In molecular electrostatic potential (MEP) map the NO2 group exhibits a blue color, indicating the presence of nucleophilic sites. The density of state (DOS), non-covalent interaction (NCI), electron localized function (ELF), localized orbital locator (LOL), Mulliken atomic charges, and reactive sites have been investigated. The anti-bacterial, antifungal, antitoxin, antiviral, and antimycobacterial studies have been investigated. The molecular docking investigations of the compound were also conducted. Using the Auto-dock program, the compound S1 molecular docking study has been conducted. The molecular docking study's lowest binding energy is -7.91, -5.62, -6.92, and -5.85 kcal/mol for 4XGK, 4ATO, 3U9 G, and 2DP4 respectively.
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
