{"title":"作为潜在抗癌剂的新型 s-三嗪衍生物:合成、DFT、DNA 结合、分子对接、MD 模拟和硅学 ADMET 分析","authors":"Alia Mushtaq, Muhammad Moazzam Naseer","doi":"10.1016/j.molstruc.2024.140558","DOIUrl":null,"url":null,"abstract":"<div><div>The ongoing challenge of cancer in modern medicine highlights the urgent need for targeted therapeutic strategies, particularly against the EGFR/PI3K/AKT/mTOR signalling pathways, which are pivotal in tumorigenesis. This study presents the design, synthesis, characterization, DNA binding studies and computational evaluation of a novel series of trisubstituted <em>s</em>-triazine derivatives <strong>3a–n</strong> as potential anticancer agents. The synthetic methodology utilized the reaction of various oxygen-containing nucleophiles with cyanuric chloride via nucleophilic aromatic substitution, yielding the corresponding target products with high yield and purity. Density Functional Theory (DFT) calculations utilizing the B3LYP level provided insights into the electronic properties of the compounds, with derivatives <strong>3f, 3n,</strong> and <strong>3l</strong> displaying the lowest energy gaps and highest electrophilicity indices, indicative of their enhanced reactivity. DNA binding results from UV–Vis absorption spectroscopy confirmed the groove mode of binding for the most potent compound <strong>3f</strong> with the binding constant (K<sub>b</sub>) value of 6.75 × 10<sup>4</sup> M<sup>−1</sup>. Molecular docking studies against DNA (PDB ID: <span><span>3EY0</span><svg><path></path></svg></span>) and EGFR (PDB ID: <span><span>6V6O</span><svg><path></path></svg></span>) revealed strong binding affinities, with compound <strong>3f</strong> (R<sub>1</sub>= ethoxy, R<sub>2</sub>= 3-CF<sub>3</sub>) exhibiting a notable binding energy of -8.9 kcal/mol and −9.1 kcal/mol respectively. Additionally, these compounds showed significant binding interactions with PI3K (PDB ID: <span><span>5HJB</span><svg><path></path></svg></span>) and mTOR (PDB ID: <span><span>4JSV</span><svg><path></path></svg></span>), suggesting their potential as dual inhibitors of the PI3K/AKT/mTOR pathway. The drug-likeness and ADMET profiles further support the therapeutic promise of compound <strong>3f</strong> bearing a 3-trifluoromethyl substituent.</div></div>","PeriodicalId":16414,"journal":{"name":"Journal of Molecular Structure","volume":"1322 ","pages":"Article 140558"},"PeriodicalIF":4.7000,"publicationDate":"2025-02-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Novel s-triazine derivatives as potential anticancer agents: Synthesis, DFT, DNA binding, molecular docking, MD simulation and in silico ADMET profiling\",\"authors\":\"Alia Mushtaq, Muhammad Moazzam Naseer\",\"doi\":\"10.1016/j.molstruc.2024.140558\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The ongoing challenge of cancer in modern medicine highlights the urgent need for targeted therapeutic strategies, particularly against the EGFR/PI3K/AKT/mTOR signalling pathways, which are pivotal in tumorigenesis. This study presents the design, synthesis, characterization, DNA binding studies and computational evaluation of a novel series of trisubstituted <em>s</em>-triazine derivatives <strong>3a–n</strong> as potential anticancer agents. The synthetic methodology utilized the reaction of various oxygen-containing nucleophiles with cyanuric chloride via nucleophilic aromatic substitution, yielding the corresponding target products with high yield and purity. Density Functional Theory (DFT) calculations utilizing the B3LYP level provided insights into the electronic properties of the compounds, with derivatives <strong>3f, 3n,</strong> and <strong>3l</strong> displaying the lowest energy gaps and highest electrophilicity indices, indicative of their enhanced reactivity. DNA binding results from UV–Vis absorption spectroscopy confirmed the groove mode of binding for the most potent compound <strong>3f</strong> with the binding constant (K<sub>b</sub>) value of 6.75 × 10<sup>4</sup> M<sup>−1</sup>. Molecular docking studies against DNA (PDB ID: <span><span>3EY0</span><svg><path></path></svg></span>) and EGFR (PDB ID: <span><span>6V6O</span><svg><path></path></svg></span>) revealed strong binding affinities, with compound <strong>3f</strong> (R<sub>1</sub>= ethoxy, R<sub>2</sub>= 3-CF<sub>3</sub>) exhibiting a notable binding energy of -8.9 kcal/mol and −9.1 kcal/mol respectively. Additionally, these compounds showed significant binding interactions with PI3K (PDB ID: <span><span>5HJB</span><svg><path></path></svg></span>) and mTOR (PDB ID: <span><span>4JSV</span><svg><path></path></svg></span>), suggesting their potential as dual inhibitors of the PI3K/AKT/mTOR pathway. The drug-likeness and ADMET profiles further support the therapeutic promise of compound <strong>3f</strong> bearing a 3-trifluoromethyl substituent.</div></div>\",\"PeriodicalId\":16414,\"journal\":{\"name\":\"Journal of Molecular Structure\",\"volume\":\"1322 \",\"pages\":\"Article 140558\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2025-02-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Structure\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022286024030667\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/2 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Structure","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022286024030667","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/2 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Novel s-triazine derivatives as potential anticancer agents: Synthesis, DFT, DNA binding, molecular docking, MD simulation and in silico ADMET profiling
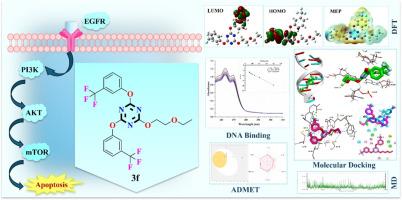
The ongoing challenge of cancer in modern medicine highlights the urgent need for targeted therapeutic strategies, particularly against the EGFR/PI3K/AKT/mTOR signalling pathways, which are pivotal in tumorigenesis. This study presents the design, synthesis, characterization, DNA binding studies and computational evaluation of a novel series of trisubstituted s-triazine derivatives 3a–n as potential anticancer agents. The synthetic methodology utilized the reaction of various oxygen-containing nucleophiles with cyanuric chloride via nucleophilic aromatic substitution, yielding the corresponding target products with high yield and purity. Density Functional Theory (DFT) calculations utilizing the B3LYP level provided insights into the electronic properties of the compounds, with derivatives 3f, 3n, and 3l displaying the lowest energy gaps and highest electrophilicity indices, indicative of their enhanced reactivity. DNA binding results from UV–Vis absorption spectroscopy confirmed the groove mode of binding for the most potent compound 3f with the binding constant (Kb) value of 6.75 × 104 M−1. Molecular docking studies against DNA (PDB ID: 3EY0) and EGFR (PDB ID: 6V6O) revealed strong binding affinities, with compound 3f (R1= ethoxy, R2= 3-CF3) exhibiting a notable binding energy of -8.9 kcal/mol and −9.1 kcal/mol respectively. Additionally, these compounds showed significant binding interactions with PI3K (PDB ID: 5HJB) and mTOR (PDB ID: 4JSV), suggesting their potential as dual inhibitors of the PI3K/AKT/mTOR pathway. The drug-likeness and ADMET profiles further support the therapeutic promise of compound 3f bearing a 3-trifluoromethyl substituent.
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
