Hanna D. Hobbs, Lowen M. Hobbs, Robert W. Zoellner
{"title":"密度泛函理论计算得出的铜氧化态极限:氟铜络合物 [CuFn]x,其中 n = 1 至 6,x = 3+ 至 5-","authors":"Hanna D. Hobbs, Lowen M. Hobbs, Robert W. Zoellner","doi":"10.1016/j.comptc.2024.114942","DOIUrl":null,"url":null,"abstract":"<div><div>Density functional theory calculations, at the ωB97X-D/6-311+G* level of theory, were performed on homoleptic fluoro-copper complexes [CuF<em><sub>n</sub></em>]<em><sup>x</sup></em>, <em>n</em> = 1 through 6 and <em>x</em> = 3+ through 5−, to determine the highest positive and lowest negative copper oxidation states that can be supported in these complexes. Only singlet and doublet spin states were investigated. All fluoro-copper stoichiometries stabilized copper(III) or greater. However, some stoichiometries stabilized oxidation states up to copper(VI), and the greatest positive copper oxidation state was copper(VIII) in the distorted octahedral [CuF<sub>6</sub>]<sup>2+</sup> cation. Oxidation states as negative as copper(–IV) in the diatomic [CuF]<sup>5−</sup> anion and copper(–III) in the triatomic [CuF<sub>2</sub>]<sup>5−</sup> were also observed as optimized minima, although no negative oxidation states were calculated to exist for fluoro-copper complexes containing more than two fluorine atoms. No singlet or doublet fluoro-copper complexes with charges more positive than 3+, more negative than 5−, or of Cu(VII), could be optimized.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114942"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The limits of copper oxidation states from density functional theory computations: Fluoro-copper complexes, [CuFn]x, where n = 1 through 6 and x = 3+ through 5−\",\"authors\":\"Hanna D. Hobbs, Lowen M. Hobbs, Robert W. Zoellner\",\"doi\":\"10.1016/j.comptc.2024.114942\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Density functional theory calculations, at the ωB97X-D/6-311+G* level of theory, were performed on homoleptic fluoro-copper complexes [CuF<em><sub>n</sub></em>]<em><sup>x</sup></em>, <em>n</em> = 1 through 6 and <em>x</em> = 3+ through 5−, to determine the highest positive and lowest negative copper oxidation states that can be supported in these complexes. Only singlet and doublet spin states were investigated. All fluoro-copper stoichiometries stabilized copper(III) or greater. However, some stoichiometries stabilized oxidation states up to copper(VI), and the greatest positive copper oxidation state was copper(VIII) in the distorted octahedral [CuF<sub>6</sub>]<sup>2+</sup> cation. Oxidation states as negative as copper(–IV) in the diatomic [CuF]<sup>5−</sup> anion and copper(–III) in the triatomic [CuF<sub>2</sub>]<sup>5−</sup> were also observed as optimized minima, although no negative oxidation states were calculated to exist for fluoro-copper complexes containing more than two fluorine atoms. No singlet or doublet fluoro-copper complexes with charges more positive than 3+, more negative than 5−, or of Cu(VII), could be optimized.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114942\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X2400481X\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/9 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X2400481X","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/9 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
The limits of copper oxidation states from density functional theory computations: Fluoro-copper complexes, [CuFn]x, where n = 1 through 6 and x = 3+ through 5−
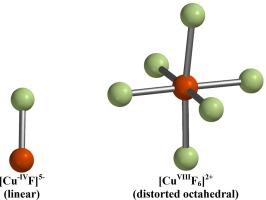
Density functional theory calculations, at the ωB97X-D/6-311+G* level of theory, were performed on homoleptic fluoro-copper complexes [CuFn]x, n = 1 through 6 and x = 3+ through 5−, to determine the highest positive and lowest negative copper oxidation states that can be supported in these complexes. Only singlet and doublet spin states were investigated. All fluoro-copper stoichiometries stabilized copper(III) or greater. However, some stoichiometries stabilized oxidation states up to copper(VI), and the greatest positive copper oxidation state was copper(VIII) in the distorted octahedral [CuF6]2+ cation. Oxidation states as negative as copper(–IV) in the diatomic [CuF]5− anion and copper(–III) in the triatomic [CuF2]5− were also observed as optimized minima, although no negative oxidation states were calculated to exist for fluoro-copper complexes containing more than two fluorine atoms. No singlet or doublet fluoro-copper complexes with charges more positive than 3+, more negative than 5−, or of Cu(VII), could be optimized.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
