Jing Yi , Tong Yan , Anqi Li , Changluo Zheng , Lidong Zhang , Longwei Cheng , Lili Lei , Pan Wang
{"title":"二乙胺脱氢及其异构化和分解反应动力学的理论和模型研究","authors":"Jing Yi , Tong Yan , Anqi Li , Changluo Zheng , Lidong Zhang , Longwei Cheng , Lili Lei , Pan Wang","doi":"10.1016/j.comptc.2024.114950","DOIUrl":null,"url":null,"abstract":"<div><div>Small-molecule nitrogen-containing contaminants can be produced by the pyrolysis of diethylamine (DEA) alone or in conjunction with other molecules. To investigate the theoretical chemistry of the combustion of DEA, the CBS method determines the potential energy surface and the rate constants are calculated based on a combination of RRKM and TST theories. The H-abstraction reactions by H radicals are more kinetically advantageous. Site α has the most difficult H-abstraction procedure, but site γ has the competitive capacity to form subsequent products. The H-abstraction by NO<sub>2</sub> radical produces <em>trans</em>-HONO, <em>cis</em>-HONO, and HNO<sub>2</sub>. For the reaction channels of DEA radicals, decomposition reactions have more kinetic advantages than isomerization. Kinetic parameters were obtained and the model was modified based on the fitted rate constants. The modified model has better predictive ability at low temperatures. This work provides extensive data to improve the modeling of DEA combustion at low and medium temperatures.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114950"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Theoretical and modeling studies on the kinetics of diethylamine dehydrogenation and subsequent isomerization and decomposition reactions\",\"authors\":\"Jing Yi , Tong Yan , Anqi Li , Changluo Zheng , Lidong Zhang , Longwei Cheng , Lili Lei , Pan Wang\",\"doi\":\"10.1016/j.comptc.2024.114950\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Small-molecule nitrogen-containing contaminants can be produced by the pyrolysis of diethylamine (DEA) alone or in conjunction with other molecules. To investigate the theoretical chemistry of the combustion of DEA, the CBS method determines the potential energy surface and the rate constants are calculated based on a combination of RRKM and TST theories. The H-abstraction reactions by H radicals are more kinetically advantageous. Site α has the most difficult H-abstraction procedure, but site γ has the competitive capacity to form subsequent products. The H-abstraction by NO<sub>2</sub> radical produces <em>trans</em>-HONO, <em>cis</em>-HONO, and HNO<sub>2</sub>. For the reaction channels of DEA radicals, decomposition reactions have more kinetic advantages than isomerization. Kinetic parameters were obtained and the model was modified based on the fitted rate constants. The modified model has better predictive ability at low temperatures. This work provides extensive data to improve the modeling of DEA combustion at low and medium temperatures.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114950\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004894\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004894","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/30 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
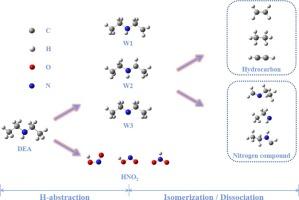
二乙胺(DEA)单独或与其他分子一起热解可产生小分子含氮污染物。为了研究二乙胺燃烧的理论化学性质,CBS 方法确定了势能面,并根据 RRKM 和 TST 理论组合计算了速率常数。通过 H 自由基进行的 H 萃取反应在动力学上更具优势。位点 α 的吸氢过程最为困难,但位点 γ 具有形成后续产物的竞争能力。NO2 自由基萃取 H 会产生反式-HONO、顺式-HONO 和 HNO2。在 DEA 自由基的反应通道中,分解反应比异构化反应具有更大的动力学优势。根据拟合的速率常数获得了动力学参数并修改了模型。修改后的模型在低温下具有更好的预测能力。这项工作为改进中低温下的 DEA 燃烧模型提供了大量数据。
Theoretical and modeling studies on the kinetics of diethylamine dehydrogenation and subsequent isomerization and decomposition reactions
Small-molecule nitrogen-containing contaminants can be produced by the pyrolysis of diethylamine (DEA) alone or in conjunction with other molecules. To investigate the theoretical chemistry of the combustion of DEA, the CBS method determines the potential energy surface and the rate constants are calculated based on a combination of RRKM and TST theories. The H-abstraction reactions by H radicals are more kinetically advantageous. Site α has the most difficult H-abstraction procedure, but site γ has the competitive capacity to form subsequent products. The H-abstraction by NO2 radical produces trans-HONO, cis-HONO, and HNO2. For the reaction channels of DEA radicals, decomposition reactions have more kinetic advantages than isomerization. Kinetic parameters were obtained and the model was modified based on the fitted rate constants. The modified model has better predictive ability at low temperatures. This work provides extensive data to improve the modeling of DEA combustion at low and medium temperatures.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
