{"title":"替代掺杂磷烯上钠吸附和扩散的第一性原理研究","authors":"Sneha Upadhyay , Pankaj Srivastava","doi":"10.1016/j.comptc.2024.114955","DOIUrl":null,"url":null,"abstract":"<div><div>Phosphorene, due to its remarkable semiconducting properties, prevailed as principal material of research for decades. The substitutional doping of carbon (C), silicon (Si), oxygen (O) and sulphur (S) atom was reported earlier. DFT studies are done towards the unexplored area of sodium (Na) adsorption and diffusion over these doped phosphorene structures. The effective adsorption energies for C-, Si-, O- and S- doped phosphorene are −3.19 eV, −2.63 eV, −3.12 eV and −2.39 eV respectively. The minimum value of activation energies are 0.68 eV, 0.45 eV, 1.00 eV and 0.26 eV respectively. Hence the diffusion and intercalation–deintercalation process seem to be supportive. The lattice integrity is maintained in the whole process. Good amount of charge transfer shows better conductivity supported by band structure and density of states diagrams. Based on these data, the proposed doped phosphorene configurations can be predicted suitable for further studies towards anode application in Na-ion battery.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114955"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"First-principles study of sodium adsorption and diffusion over substitutionally doped phosphorene\",\"authors\":\"Sneha Upadhyay , Pankaj Srivastava\",\"doi\":\"10.1016/j.comptc.2024.114955\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Phosphorene, due to its remarkable semiconducting properties, prevailed as principal material of research for decades. The substitutional doping of carbon (C), silicon (Si), oxygen (O) and sulphur (S) atom was reported earlier. DFT studies are done towards the unexplored area of sodium (Na) adsorption and diffusion over these doped phosphorene structures. The effective adsorption energies for C-, Si-, O- and S- doped phosphorene are −3.19 eV, −2.63 eV, −3.12 eV and −2.39 eV respectively. The minimum value of activation energies are 0.68 eV, 0.45 eV, 1.00 eV and 0.26 eV respectively. Hence the diffusion and intercalation–deintercalation process seem to be supportive. The lattice integrity is maintained in the whole process. Good amount of charge transfer shows better conductivity supported by band structure and density of states diagrams. Based on these data, the proposed doped phosphorene configurations can be predicted suitable for further studies towards anode application in Na-ion battery.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114955\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004948\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/6 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004948","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/6 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
First-principles study of sodium adsorption and diffusion over substitutionally doped phosphorene
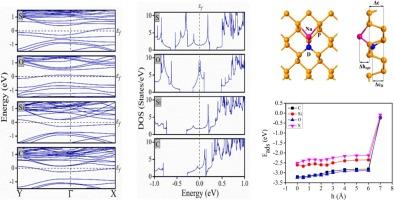
Phosphorene, due to its remarkable semiconducting properties, prevailed as principal material of research for decades. The substitutional doping of carbon (C), silicon (Si), oxygen (O) and sulphur (S) atom was reported earlier. DFT studies are done towards the unexplored area of sodium (Na) adsorption and diffusion over these doped phosphorene structures. The effective adsorption energies for C-, Si-, O- and S- doped phosphorene are −3.19 eV, −2.63 eV, −3.12 eV and −2.39 eV respectively. The minimum value of activation energies are 0.68 eV, 0.45 eV, 1.00 eV and 0.26 eV respectively. Hence the diffusion and intercalation–deintercalation process seem to be supportive. The lattice integrity is maintained in the whole process. Good amount of charge transfer shows better conductivity supported by band structure and density of states diagrams. Based on these data, the proposed doped phosphorene configurations can be predicted suitable for further studies towards anode application in Na-ion battery.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
