Rim Bechaieb , Maha F. El-Tohamy , Gamal A.E. Mostafa
{"title":"氯锡酸(II)杂化材料中的卤素取代效应:对 2(C4H4FN3O)-SnX6-2(H2O)(X = F、Cl、Br、I)的 DFT 研究的启示","authors":"Rim Bechaieb , Maha F. El-Tohamy , Gamal A.E. Mostafa","doi":"10.1016/j.comptc.2024.114978","DOIUrl":null,"url":null,"abstract":"<div><div>The development of new materials is crucial for advancing technologies in material science, energy, and biology. In this study, we investigate the effect of Halogen-substitution on the compound 2(C<sub>4</sub>H<sub>4</sub>FN<sub>3</sub>O)·SnX<sub>6</sub>·2(H<sub>2</sub>O) (where X = F, Cl, Br, and I). Using extensive theoretical analysis, we examined the impact of halogen substitution on the structural properties, band gap energy (Eg), and chemical behavior. Our results show that the Eg decreases with the increasing atomic number of halogens. The I-substituted compound is the most reactive and exhibits significant intramolecular charge transfer, enhancing its antioxidant properties. The spatial distribution of electron density in the Frontier Molecular Orbitals (FMOs) indicates that the HOMO is localized on the inorganic anion and the LUMO on the organic cation, a pattern consistent across all substituted compounds. These findings highlight the potential of these compounds in electron transfer reactions and provide insights for tailoring them for specific chemical and biochemical applications.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114978"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Halogen substitution effects in Chlorostannate(II) hybrid material: Insights from DFT study on 2(C4H4FN3O)·SnX6·2(H2O), (X = F, Cl, Br, I)\",\"authors\":\"Rim Bechaieb , Maha F. El-Tohamy , Gamal A.E. Mostafa\",\"doi\":\"10.1016/j.comptc.2024.114978\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The development of new materials is crucial for advancing technologies in material science, energy, and biology. In this study, we investigate the effect of Halogen-substitution on the compound 2(C<sub>4</sub>H<sub>4</sub>FN<sub>3</sub>O)·SnX<sub>6</sub>·2(H<sub>2</sub>O) (where X = F, Cl, Br, and I). Using extensive theoretical analysis, we examined the impact of halogen substitution on the structural properties, band gap energy (Eg), and chemical behavior. Our results show that the Eg decreases with the increasing atomic number of halogens. The I-substituted compound is the most reactive and exhibits significant intramolecular charge transfer, enhancing its antioxidant properties. The spatial distribution of electron density in the Frontier Molecular Orbitals (FMOs) indicates that the HOMO is localized on the inorganic anion and the LUMO on the organic cation, a pattern consistent across all substituted compounds. These findings highlight the potential of these compounds in electron transfer reactions and provide insights for tailoring them for specific chemical and biochemical applications.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114978\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24005176\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24005176","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/5 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Halogen substitution effects in Chlorostannate(II) hybrid material: Insights from DFT study on 2(C4H4FN3O)·SnX6·2(H2O), (X = F, Cl, Br, I)
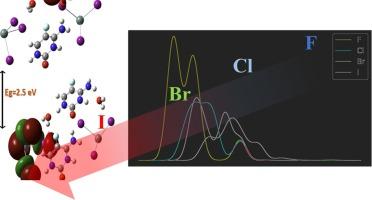
The development of new materials is crucial for advancing technologies in material science, energy, and biology. In this study, we investigate the effect of Halogen-substitution on the compound 2(C4H4FN3O)·SnX6·2(H2O) (where X = F, Cl, Br, and I). Using extensive theoretical analysis, we examined the impact of halogen substitution on the structural properties, band gap energy (Eg), and chemical behavior. Our results show that the Eg decreases with the increasing atomic number of halogens. The I-substituted compound is the most reactive and exhibits significant intramolecular charge transfer, enhancing its antioxidant properties. The spatial distribution of electron density in the Frontier Molecular Orbitals (FMOs) indicates that the HOMO is localized on the inorganic anion and the LUMO on the organic cation, a pattern consistent across all substituted compounds. These findings highlight the potential of these compounds in electron transfer reactions and provide insights for tailoring them for specific chemical and biochemical applications.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
