{"title":"通过基于密度的基集修正实现化学精确量子计算的捷径","authors":"Diata Traore, Olivier Adjoua, César Feniou, Ioanna-Maria Lygatsika, Yvon Maday, Evgeny Posenitskiy, Kerstin Hammernik, Alberto Peruzzo, Julien Toulouse, Emmanuel Giner, Jean-Philip Piquemal","doi":"10.1038/s42004-024-01348-3","DOIUrl":null,"url":null,"abstract":"Using GPU-accelerated state-vector emulation, we propose to embed a quantum computing ansatz into density-functional theory via density-based basis-set corrections to obtain quantitative quantum-chemistry results on molecules that would otherwise require brute-force quantum calculations using hundreds of logical qubits. Indeed, accessing a quantitative description of chemical systems while minimizing quantum resources is an essential challenge given the limited qubit capabilities of current quantum processors. We provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget. The resulting approach self-consistently accelerates the basis-set convergence, improving electronic densities, ground-state energies, and first-order properties (e.g. dipole moments), but can also serve as a classical, a posteriori, energy correction to quantum hardware calculations with expected applications in drug design and materials science. Quantum computing offers a promising approach to solving electronic-structure problems, but a quantitative description of chemical systems while minimizing computing resources is an essential challenge. Here, the authors provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget.","PeriodicalId":10529,"journal":{"name":"Communications Chemistry","volume":" ","pages":"1-13"},"PeriodicalIF":6.2000,"publicationDate":"2024-11-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s42004-024-01348-3.pdf","citationCount":"0","resultStr":"{\"title\":\"Shortcut to chemically accurate quantum computing via density-based basis-set correction\",\"authors\":\"Diata Traore, Olivier Adjoua, César Feniou, Ioanna-Maria Lygatsika, Yvon Maday, Evgeny Posenitskiy, Kerstin Hammernik, Alberto Peruzzo, Julien Toulouse, Emmanuel Giner, Jean-Philip Piquemal\",\"doi\":\"10.1038/s42004-024-01348-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Using GPU-accelerated state-vector emulation, we propose to embed a quantum computing ansatz into density-functional theory via density-based basis-set corrections to obtain quantitative quantum-chemistry results on molecules that would otherwise require brute-force quantum calculations using hundreds of logical qubits. Indeed, accessing a quantitative description of chemical systems while minimizing quantum resources is an essential challenge given the limited qubit capabilities of current quantum processors. We provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget. The resulting approach self-consistently accelerates the basis-set convergence, improving electronic densities, ground-state energies, and first-order properties (e.g. dipole moments), but can also serve as a classical, a posteriori, energy correction to quantum hardware calculations with expected applications in drug design and materials science. Quantum computing offers a promising approach to solving electronic-structure problems, but a quantitative description of chemical systems while minimizing computing resources is an essential challenge. Here, the authors provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget.\",\"PeriodicalId\":10529,\"journal\":{\"name\":\"Communications Chemistry\",\"volume\":\" \",\"pages\":\"1-13\"},\"PeriodicalIF\":6.2000,\"publicationDate\":\"2024-11-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s42004-024-01348-3.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Communications Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.nature.com/articles/s42004-024-01348-3\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Communications Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.nature.com/articles/s42004-024-01348-3","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Shortcut to chemically accurate quantum computing via density-based basis-set correction
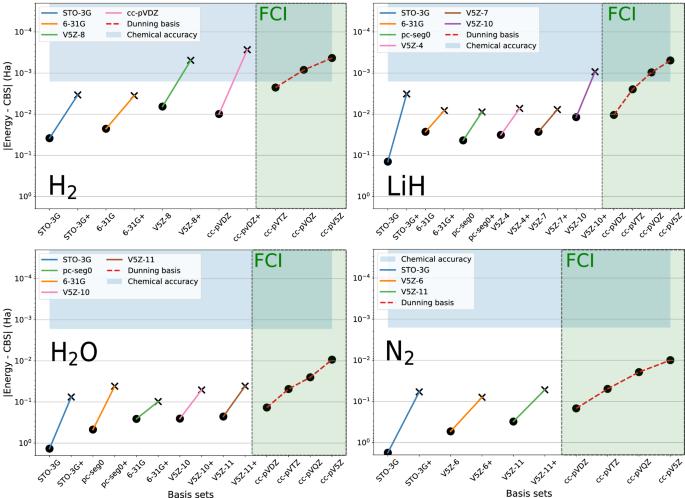
Using GPU-accelerated state-vector emulation, we propose to embed a quantum computing ansatz into density-functional theory via density-based basis-set corrections to obtain quantitative quantum-chemistry results on molecules that would otherwise require brute-force quantum calculations using hundreds of logical qubits. Indeed, accessing a quantitative description of chemical systems while minimizing quantum resources is an essential challenge given the limited qubit capabilities of current quantum processors. We provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget. The resulting approach self-consistently accelerates the basis-set convergence, improving electronic densities, ground-state energies, and first-order properties (e.g. dipole moments), but can also serve as a classical, a posteriori, energy correction to quantum hardware calculations with expected applications in drug design and materials science. Quantum computing offers a promising approach to solving electronic-structure problems, but a quantitative description of chemical systems while minimizing computing resources is an essential challenge. Here, the authors provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget.
期刊介绍:
Communications Chemistry is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the chemical sciences. Research papers published by the journal represent significant advances bringing new chemical insight to a specialized area of research. We also aim to provide a community forum for issues of importance to all chemists, regardless of sub-discipline.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
