Yaling Zhang, Chaoqin Cao, Yuanbin She, Huw M L Davies, Yun-Fang Yang, K N Houk
{"title":"Davies Ambimodal C-H Functionalization/Cope Rearrangement Reaction 的分子动力学。","authors":"Yaling Zhang, Chaoqin Cao, Yuanbin She, Huw M L Davies, Yun-Fang Yang, K N Houk","doi":"10.1021/acs.joc.4c01682","DOIUrl":null,"url":null,"abstract":"<p><p>The mechanism of the dirhodium-catalyzed combined C-H functionalization/Cope rearrangement (CH/Cope) reaction discovered by the Davies group has been investigated with density functional theory (DFT) calculations and quasi-classical molecular dynamics (MD) simulations. Computations from the Davies group previously showed that there is a post-transition state bifurcation leading to a direct CH reaction and also to the CH/Cope product. While this work was in preparation, the Tantillo group and the Ess group independently reported quantum mechanical and molecular dynamics studies on the dirhodium-tetracarboxylate-catalyzed diazoester CH/Cope and CH insertion reactions with 1,3-cyclohexadiene and 1,4-cyclohexadiene, respectively. The Tantillo group cited \"dynamic mismatching\" to explain the origins of the low yield of CH/Cope products in some experiments; the Ess group explained the origins of product selectivity from the perspective of TS vibrational modes and their synchronization that occurs at the entropic intermediates. We report quasi-classical trajectories for the reaction of the carbene with 1-methylcyclohexene that afford both the CH/Cope and C-H insertion products. After passing through the transition state that involves mostly hydrogen transfer, momentum drives the reaction trajectories toward the CH/Cope products.</p>","PeriodicalId":57,"journal":{"name":"Journal of Organic Chemistry","volume":" ","pages":"17176-17186"},"PeriodicalIF":3.6000,"publicationDate":"2024-12-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12442840/pdf/","citationCount":"0","resultStr":"{\"title\":\"Molecular Dynamics of the Davies Ambimodal C-H Functionalization/Cope Rearrangement Reaction.\",\"authors\":\"Yaling Zhang, Chaoqin Cao, Yuanbin She, Huw M L Davies, Yun-Fang Yang, K N Houk\",\"doi\":\"10.1021/acs.joc.4c01682\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The mechanism of the dirhodium-catalyzed combined C-H functionalization/Cope rearrangement (CH/Cope) reaction discovered by the Davies group has been investigated with density functional theory (DFT) calculations and quasi-classical molecular dynamics (MD) simulations. Computations from the Davies group previously showed that there is a post-transition state bifurcation leading to a direct CH reaction and also to the CH/Cope product. While this work was in preparation, the Tantillo group and the Ess group independently reported quantum mechanical and molecular dynamics studies on the dirhodium-tetracarboxylate-catalyzed diazoester CH/Cope and CH insertion reactions with 1,3-cyclohexadiene and 1,4-cyclohexadiene, respectively. The Tantillo group cited \\\"dynamic mismatching\\\" to explain the origins of the low yield of CH/Cope products in some experiments; the Ess group explained the origins of product selectivity from the perspective of TS vibrational modes and their synchronization that occurs at the entropic intermediates. We report quasi-classical trajectories for the reaction of the carbene with 1-methylcyclohexene that afford both the CH/Cope and C-H insertion products. After passing through the transition state that involves mostly hydrogen transfer, momentum drives the reaction trajectories toward the CH/Cope products.</p>\",\"PeriodicalId\":57,\"journal\":{\"name\":\"Journal of Organic Chemistry\",\"volume\":\" \",\"pages\":\"17176-17186\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-12-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12442840/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Organic Chemistry\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.joc.4c01682\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/19 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Organic Chemistry","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.joc.4c01682","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/19 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
Molecular Dynamics of the Davies Ambimodal C-H Functionalization/Cope Rearrangement Reaction.
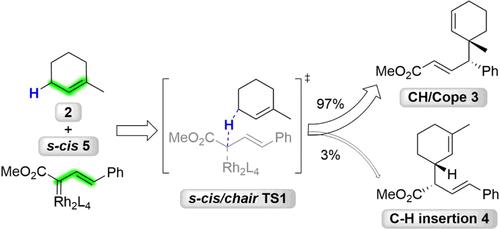
The mechanism of the dirhodium-catalyzed combined C-H functionalization/Cope rearrangement (CH/Cope) reaction discovered by the Davies group has been investigated with density functional theory (DFT) calculations and quasi-classical molecular dynamics (MD) simulations. Computations from the Davies group previously showed that there is a post-transition state bifurcation leading to a direct CH reaction and also to the CH/Cope product. While this work was in preparation, the Tantillo group and the Ess group independently reported quantum mechanical and molecular dynamics studies on the dirhodium-tetracarboxylate-catalyzed diazoester CH/Cope and CH insertion reactions with 1,3-cyclohexadiene and 1,4-cyclohexadiene, respectively. The Tantillo group cited "dynamic mismatching" to explain the origins of the low yield of CH/Cope products in some experiments; the Ess group explained the origins of product selectivity from the perspective of TS vibrational modes and their synchronization that occurs at the entropic intermediates. We report quasi-classical trajectories for the reaction of the carbene with 1-methylcyclohexene that afford both the CH/Cope and C-H insertion products. After passing through the transition state that involves mostly hydrogen transfer, momentum drives the reaction trajectories toward the CH/Cope products.
期刊介绍:
Journal of Organic Chemistry welcomes original contributions of fundamental research in all branches of the theory and practice of organic chemistry. In selecting manuscripts for publication, the editors place emphasis on the quality and novelty of the work, as well as the breadth of interest to the organic chemistry community.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
