{"title":"8 名鸟氨酸转氨酶缺乏症患儿的临床和基因分析:两种新型突变。","authors":"Chen Zhang, Junli Shan, Jiaqi Su, Guan Wang, Qin Huo, Rui Xu, Meng Dong","doi":"10.1212/NXG.0000000000200204","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Cases and studies of neurologic symptoms in children caused by genetic metabolic diseases have been widely reported. Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle disorder, which is due to mutations in the OTC gene located on chromosome Xp21.1. In this study, we analyzed the clinical and genetic characteristics of 8 Chinese children diagnosed with OTCD.</p><p><strong>Methods: </strong>A total of 8 patients (5 male and 3 female) were diagnosed with OTCD by biochemical and molecular analysis between 2015 and 2023. Clinical manifestations, biochemical features, and OTC gene sequencing analysis were reviewed retrospectively. The effect of c.664-1G>C on OTC mRNA synthesis was confirmed by a minigene splicing assay.</p><p><strong>Results: </strong>All children were late-onset patients, with a median onset age of 3.6 years (range 1.8-17 years). Neurologic symptoms caused by hyperammonemia include vomiting, coma, dyssomnia, and seizures. The peak plasma ammonia levels ranged from 149 to 4,490 μmol/L, and alanine transaminase levels ranged from 20 to 1316 U/L. Four of them had received CRRT, and only 1 patient was admitted for liver transplantation. By December 2023, 4 patients had survived and 4 were deceased. Blood amino acids or urinary organic acids were detected in 7 cases. All patients underwent whole-exome sequencing, and 2 novel mutations were revealed (P1, c.617dupT and P2, c.664-1G>C). The alteration (c.664-1G>C) leads to the deletion of a 54-bp sequence in the exon 7 of the OTC gene (NM_000531.6: c.664_717del).</p><p><strong>Discussion: </strong>Two novel pathogenic variants in the OTC gene were confirmed in 8 Chinese children by biochemical findings and genetic analysis. These findings will provide a better understanding of the diagnosis and treatment of pediatric patients with OTCD.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"10 6","pages":"e200204"},"PeriodicalIF":3.3000,"publicationDate":"2024-11-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11573262/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical and Genetic Analysis of 8 Children With Ornithine Transcarbamylase Deficiency: Two Novel Mutations.\",\"authors\":\"Chen Zhang, Junli Shan, Jiaqi Su, Guan Wang, Qin Huo, Rui Xu, Meng Dong\",\"doi\":\"10.1212/NXG.0000000000200204\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objectives: </strong>Cases and studies of neurologic symptoms in children caused by genetic metabolic diseases have been widely reported. Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle disorder, which is due to mutations in the OTC gene located on chromosome Xp21.1. In this study, we analyzed the clinical and genetic characteristics of 8 Chinese children diagnosed with OTCD.</p><p><strong>Methods: </strong>A total of 8 patients (5 male and 3 female) were diagnosed with OTCD by biochemical and molecular analysis between 2015 and 2023. Clinical manifestations, biochemical features, and OTC gene sequencing analysis were reviewed retrospectively. The effect of c.664-1G>C on OTC mRNA synthesis was confirmed by a minigene splicing assay.</p><p><strong>Results: </strong>All children were late-onset patients, with a median onset age of 3.6 years (range 1.8-17 years). Neurologic symptoms caused by hyperammonemia include vomiting, coma, dyssomnia, and seizures. The peak plasma ammonia levels ranged from 149 to 4,490 μmol/L, and alanine transaminase levels ranged from 20 to 1316 U/L. Four of them had received CRRT, and only 1 patient was admitted for liver transplantation. By December 2023, 4 patients had survived and 4 were deceased. Blood amino acids or urinary organic acids were detected in 7 cases. All patients underwent whole-exome sequencing, and 2 novel mutations were revealed (P1, c.617dupT and P2, c.664-1G>C). The alteration (c.664-1G>C) leads to the deletion of a 54-bp sequence in the exon 7 of the OTC gene (NM_000531.6: c.664_717del).</p><p><strong>Discussion: </strong>Two novel pathogenic variants in the OTC gene were confirmed in 8 Chinese children by biochemical findings and genetic analysis. These findings will provide a better understanding of the diagnosis and treatment of pediatric patients with OTCD.</p>\",\"PeriodicalId\":48613,\"journal\":{\"name\":\"Neurology-Genetics\",\"volume\":\"10 6\",\"pages\":\"e200204\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-11-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11573262/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology-Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1212/NXG.0000000000200204\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200204","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Clinical and Genetic Analysis of 8 Children With Ornithine Transcarbamylase Deficiency: Two Novel Mutations.
Background and objectives: Cases and studies of neurologic symptoms in children caused by genetic metabolic diseases have been widely reported. Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle disorder, which is due to mutations in the OTC gene located on chromosome Xp21.1. In this study, we analyzed the clinical and genetic characteristics of 8 Chinese children diagnosed with OTCD.
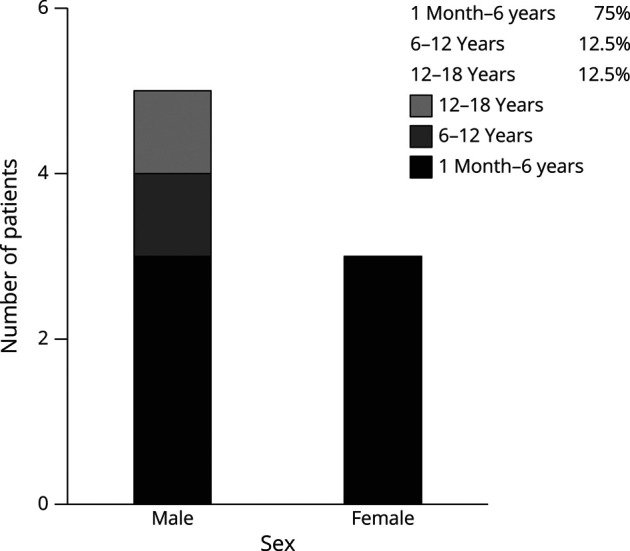
Methods: A total of 8 patients (5 male and 3 female) were diagnosed with OTCD by biochemical and molecular analysis between 2015 and 2023. Clinical manifestations, biochemical features, and OTC gene sequencing analysis were reviewed retrospectively. The effect of c.664-1G>C on OTC mRNA synthesis was confirmed by a minigene splicing assay.
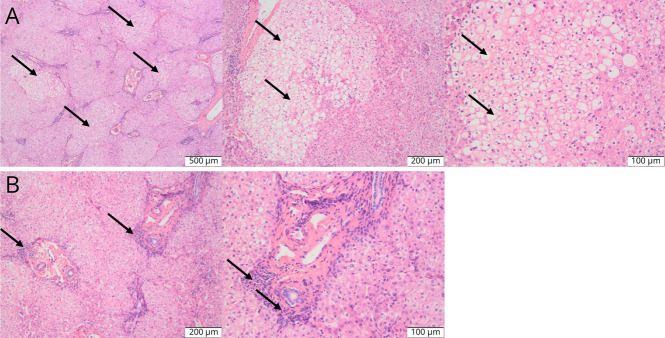
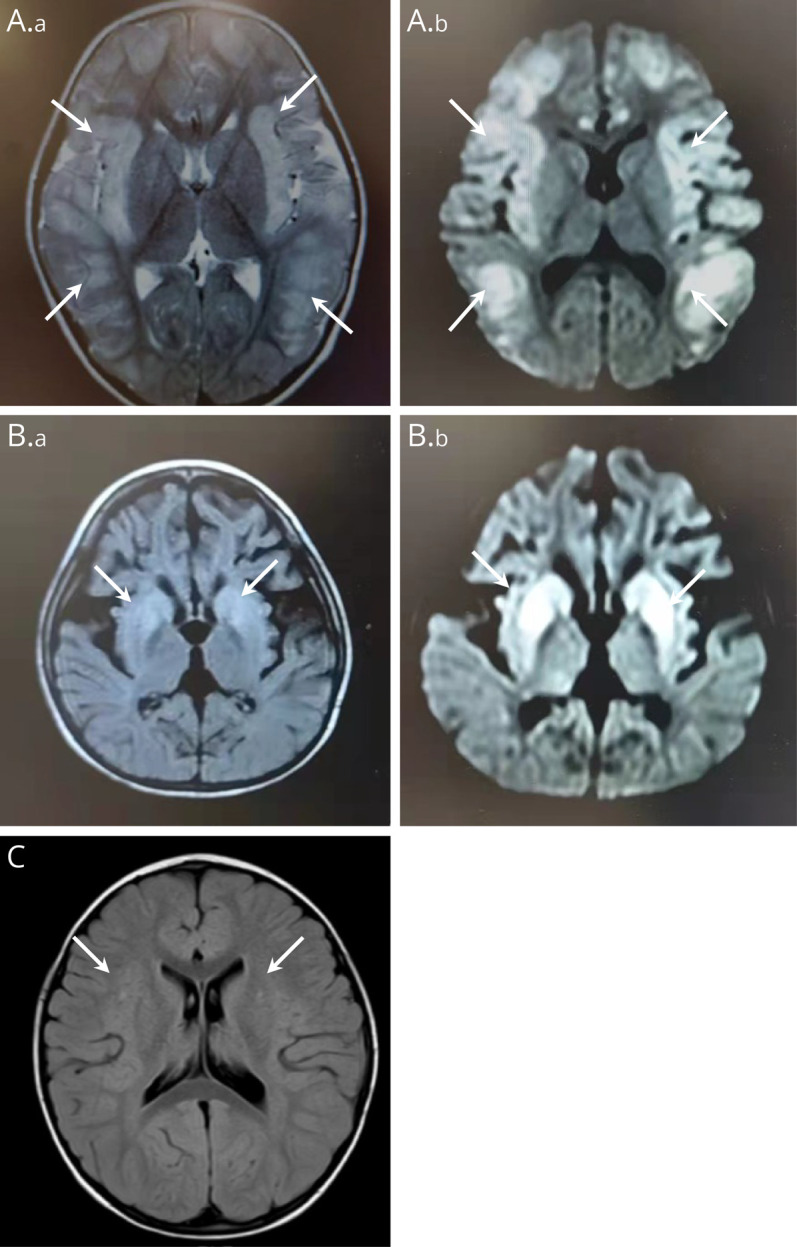
Results: All children were late-onset patients, with a median onset age of 3.6 years (range 1.8-17 years). Neurologic symptoms caused by hyperammonemia include vomiting, coma, dyssomnia, and seizures. The peak plasma ammonia levels ranged from 149 to 4,490 μmol/L, and alanine transaminase levels ranged from 20 to 1316 U/L. Four of them had received CRRT, and only 1 patient was admitted for liver transplantation. By December 2023, 4 patients had survived and 4 were deceased. Blood amino acids or urinary organic acids were detected in 7 cases. All patients underwent whole-exome sequencing, and 2 novel mutations were revealed (P1, c.617dupT and P2, c.664-1G>C). The alteration (c.664-1G>C) leads to the deletion of a 54-bp sequence in the exon 7 of the OTC gene (NM_000531.6: c.664_717del).
Discussion: Two novel pathogenic variants in the OTC gene were confirmed in 8 Chinese children by biochemical findings and genetic analysis. These findings will provide a better understanding of the diagnosis and treatment of pediatric patients with OTCD.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
