Abderrahim Ait Ouchaoui, Salah Eddine El Hadad, Marouane Aherkou, Elkamili Fadoua, Mkamel Mouad, Youssef Ramli, Anass Kettani, Ilhame Bourais
{"title":"揭示苯并三嗪的潜力:通过分子对接、动态模拟和 ADMET 分析研究 PD-L1 抑制剂在肿瘤免疫逃避中的作用的计算方法。","authors":"Abderrahim Ait Ouchaoui, Salah Eddine El Hadad, Marouane Aherkou, Elkamili Fadoua, Mkamel Mouad, Youssef Ramli, Anass Kettani, Ilhame Bourais","doi":"10.1177/11779322241298591","DOIUrl":null,"url":null,"abstract":"<p><p>The interaction between programmed cell death protein 1 (PD-1) and its ligand PD-L1 plays a crucial role in tumor immune evasion, presenting a critical target for cancer immunotherapy. Despite being effective, current monoclonal antibodies present some drawbacks such as high costs, toxicity, and resistance development. Therefore, the development of small-molecule inhibitors is necessary, especially those derived from natural sources. In this study, benzosampangine is predicted as a promising PD-L1 inhibitor, with potential applications in cancer immunotherapy. Utilizing the high-resolution crystal structure of human PD-L1 (PDB ID: 5O45), we screened 511 natural compounds, identifying benzosampangine as a top candidate with exceptional inhibitory properties. Molecular docking predicted that benzosampangine exhibits a strong binding affinity for PD-L1 (-9.4 kcal/mol) compared with established controls such as CA-170 (-6.5 kcal/mol), BMS-202 (-8.6 kcal/mol), and pyrvinium (-8.9 kcal/mol). The compound's predicted binding efficacy is highlighted by robust interactions with key amino acids (ILE54, TYR56, GLN66, MET115, ILE116, SER117, ALA121, ASP122) within the active site, notably forming 3 Pi-sulfur interactions with MET115-an interaction absents in control inhibitors. In addition, ADMET profiling suggests that over the control molecules, benzosampangine has several key advantages, including favorable solubility, permeability, metabolic stability, and low toxicity, while adhering to Lipinski's rule of five. Molecular dynamic simulations predict the stability of the benzosampangine-PD-L1 complex, reinforcing its potential to sustain inhibition of the PD-1/PD-L1 pathway. MMGBSA analysis calculated a binding free energy (ΔGbind) of -39.39 kcal/mol for the benzosampangine-PD-L1 complex, with significant contributions from Coulombic, lipophilic, and Van der Waals interactions, validating the predicted docking results. This study investigates in silico benzosampangine, predicting its better molecular interactions and pharmacokinetic profile compared with several already known PD-L1 inhibitors.</p>","PeriodicalId":9065,"journal":{"name":"Bioinformatics and Biology Insights","volume":"18 ","pages":"11779322241298591"},"PeriodicalIF":2.4000,"publicationDate":"2024-11-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11574905/pdf/","citationCount":"0","resultStr":"{\"title\":\"Unlocking Benzosampangine's Potential: A Computational Approach to Investigating, Its Role as a PD-L1 Inhibitor in Tumor Immune Evasion via Molecular Docking, Dynamic Simulation, and ADMET Profiling.\",\"authors\":\"Abderrahim Ait Ouchaoui, Salah Eddine El Hadad, Marouane Aherkou, Elkamili Fadoua, Mkamel Mouad, Youssef Ramli, Anass Kettani, Ilhame Bourais\",\"doi\":\"10.1177/11779322241298591\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The interaction between programmed cell death protein 1 (PD-1) and its ligand PD-L1 plays a crucial role in tumor immune evasion, presenting a critical target for cancer immunotherapy. Despite being effective, current monoclonal antibodies present some drawbacks such as high costs, toxicity, and resistance development. Therefore, the development of small-molecule inhibitors is necessary, especially those derived from natural sources. In this study, benzosampangine is predicted as a promising PD-L1 inhibitor, with potential applications in cancer immunotherapy. Utilizing the high-resolution crystal structure of human PD-L1 (PDB ID: 5O45), we screened 511 natural compounds, identifying benzosampangine as a top candidate with exceptional inhibitory properties. Molecular docking predicted that benzosampangine exhibits a strong binding affinity for PD-L1 (-9.4 kcal/mol) compared with established controls such as CA-170 (-6.5 kcal/mol), BMS-202 (-8.6 kcal/mol), and pyrvinium (-8.9 kcal/mol). The compound's predicted binding efficacy is highlighted by robust interactions with key amino acids (ILE54, TYR56, GLN66, MET115, ILE116, SER117, ALA121, ASP122) within the active site, notably forming 3 Pi-sulfur interactions with MET115-an interaction absents in control inhibitors. In addition, ADMET profiling suggests that over the control molecules, benzosampangine has several key advantages, including favorable solubility, permeability, metabolic stability, and low toxicity, while adhering to Lipinski's rule of five. Molecular dynamic simulations predict the stability of the benzosampangine-PD-L1 complex, reinforcing its potential to sustain inhibition of the PD-1/PD-L1 pathway. MMGBSA analysis calculated a binding free energy (ΔGbind) of -39.39 kcal/mol for the benzosampangine-PD-L1 complex, with significant contributions from Coulombic, lipophilic, and Van der Waals interactions, validating the predicted docking results. This study investigates in silico benzosampangine, predicting its better molecular interactions and pharmacokinetic profile compared with several already known PD-L1 inhibitors.</p>\",\"PeriodicalId\":9065,\"journal\":{\"name\":\"Bioinformatics and Biology Insights\",\"volume\":\"18 \",\"pages\":\"11779322241298591\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2024-11-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11574905/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics and Biology Insights\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11779322241298591\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics and Biology Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11779322241298591","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Unlocking Benzosampangine's Potential: A Computational Approach to Investigating, Its Role as a PD-L1 Inhibitor in Tumor Immune Evasion via Molecular Docking, Dynamic Simulation, and ADMET Profiling.
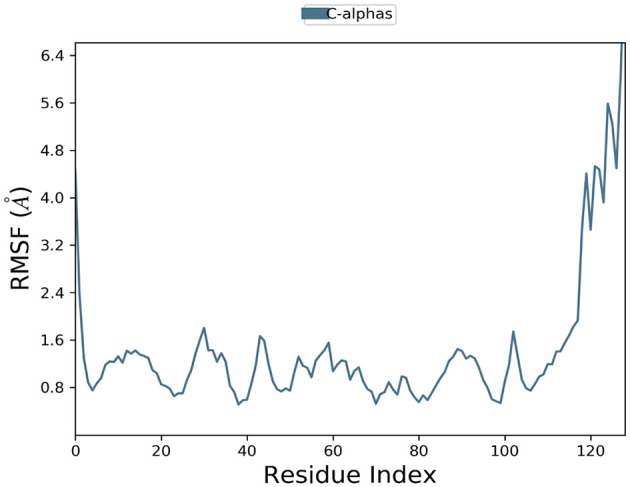
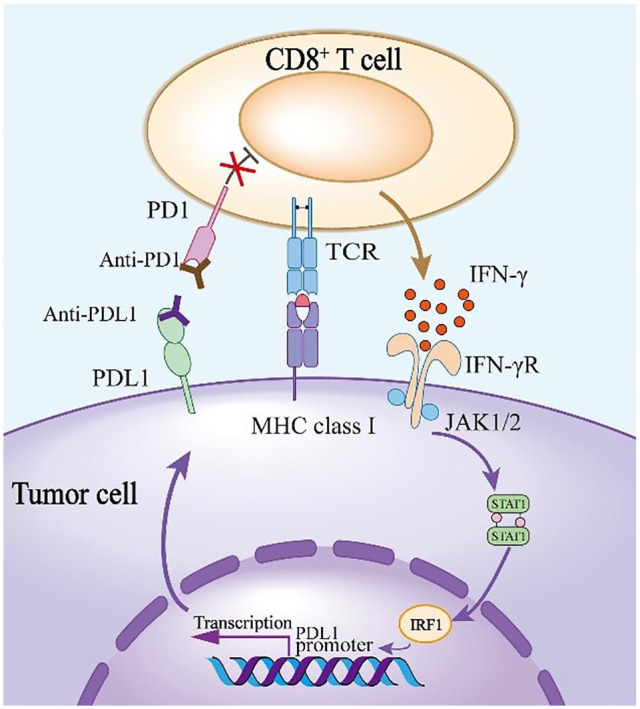
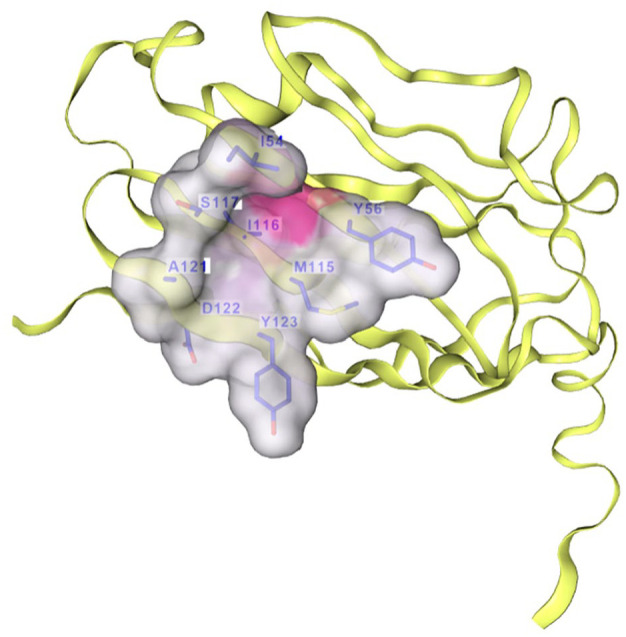
The interaction between programmed cell death protein 1 (PD-1) and its ligand PD-L1 plays a crucial role in tumor immune evasion, presenting a critical target for cancer immunotherapy. Despite being effective, current monoclonal antibodies present some drawbacks such as high costs, toxicity, and resistance development. Therefore, the development of small-molecule inhibitors is necessary, especially those derived from natural sources. In this study, benzosampangine is predicted as a promising PD-L1 inhibitor, with potential applications in cancer immunotherapy. Utilizing the high-resolution crystal structure of human PD-L1 (PDB ID: 5O45), we screened 511 natural compounds, identifying benzosampangine as a top candidate with exceptional inhibitory properties. Molecular docking predicted that benzosampangine exhibits a strong binding affinity for PD-L1 (-9.4 kcal/mol) compared with established controls such as CA-170 (-6.5 kcal/mol), BMS-202 (-8.6 kcal/mol), and pyrvinium (-8.9 kcal/mol). The compound's predicted binding efficacy is highlighted by robust interactions with key amino acids (ILE54, TYR56, GLN66, MET115, ILE116, SER117, ALA121, ASP122) within the active site, notably forming 3 Pi-sulfur interactions with MET115-an interaction absents in control inhibitors. In addition, ADMET profiling suggests that over the control molecules, benzosampangine has several key advantages, including favorable solubility, permeability, metabolic stability, and low toxicity, while adhering to Lipinski's rule of five. Molecular dynamic simulations predict the stability of the benzosampangine-PD-L1 complex, reinforcing its potential to sustain inhibition of the PD-1/PD-L1 pathway. MMGBSA analysis calculated a binding free energy (ΔGbind) of -39.39 kcal/mol for the benzosampangine-PD-L1 complex, with significant contributions from Coulombic, lipophilic, and Van der Waals interactions, validating the predicted docking results. This study investigates in silico benzosampangine, predicting its better molecular interactions and pharmacokinetic profile compared with several already known PD-L1 inhibitors.
期刊介绍:
Bioinformatics and Biology Insights is an open access, peer-reviewed journal that considers articles on bioinformatics methods and their applications which must pertain to biological insights. All papers should be easily amenable to biologists and as such help bridge the gap between theories and applications.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
