{"title":"通过三活性火山图探究单原子催化剂利用 H2O2 氧化 NO 的催化活性","authors":"Weijie Yang, Xiang Li, Ruiyang Shi, Mingye Huang, Liugang Chen, Yixiao Sun, Yanfeng Liu, Zhengyang Gao","doi":"10.1021/acs.jpcc.4c06149","DOIUrl":null,"url":null,"abstract":"The recently emerging single-atom catalysts (SACs) are proposed as promising candidates for the catalytic oxidation of NO in the field of NO pollutant reduction from coal-fired power stations. However, due to the lack of theoretical model guidance, achieving efficient catalytic oxidation of NO at room temperature still remains a major concern. Thus, in this study, the reaction pathways for the catalytic oxidation of NO by OOH radicals on eight TM–N<sub>4</sub>–C (TM = Sc, Cr, Mn, Fe, Co, Ni, Cu, and Zn) were investigated based on density functional theory calculations. Based on the Bronsted–Evans–Polanyi linear relationship and microkinetic simulations, the activity volcano plot model for the catalytic oxidation of NO by OOH has been successfully established and validated. The Fe–N<sub>4</sub>–C and Mn–N<sub>4</sub>–C catalysts showed higher reactivities among these catalysts. The energy barriers for the rate-determining steps of these two catalysts were 0.14 and 0.30 eV, respectively, illustrating that catalytic oxidation of NO is feasible at room temperature. Significantly, a tri-activity volcano plot was constructed based on the unified activity descriptor of O adsorption energy for guiding the design of SACs in H<sub>2</sub>O<sub>2</sub> catalytic oxidation of NO. The pathway of catalytic oxidation NO by OOH radicals is the dominant route in the system of SACs for the catalytic oxidation of NO by H<sub>2</sub>O<sub>2</sub>. The analysis of Bader charge and electronegativity revealed a linear correlation between the structural properties of the catalysts and the catalytic activity descriptor, which can be used to quickly predict the reactivity of SACs with different coordination environments. This work provides a new pathway for the current NO oxidation and guides future work on catalyst screening and experimental preparation.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"29 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Probing into the Catalytic Activity of Single-Atom Catalysts for NO Oxidation by H2O2 via the Tri-activity Volcano Plot\",\"authors\":\"Weijie Yang, Xiang Li, Ruiyang Shi, Mingye Huang, Liugang Chen, Yixiao Sun, Yanfeng Liu, Zhengyang Gao\",\"doi\":\"10.1021/acs.jpcc.4c06149\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The recently emerging single-atom catalysts (SACs) are proposed as promising candidates for the catalytic oxidation of NO in the field of NO pollutant reduction from coal-fired power stations. However, due to the lack of theoretical model guidance, achieving efficient catalytic oxidation of NO at room temperature still remains a major concern. Thus, in this study, the reaction pathways for the catalytic oxidation of NO by OOH radicals on eight TM–N<sub>4</sub>–C (TM = Sc, Cr, Mn, Fe, Co, Ni, Cu, and Zn) were investigated based on density functional theory calculations. Based on the Bronsted–Evans–Polanyi linear relationship and microkinetic simulations, the activity volcano plot model for the catalytic oxidation of NO by OOH has been successfully established and validated. The Fe–N<sub>4</sub>–C and Mn–N<sub>4</sub>–C catalysts showed higher reactivities among these catalysts. The energy barriers for the rate-determining steps of these two catalysts were 0.14 and 0.30 eV, respectively, illustrating that catalytic oxidation of NO is feasible at room temperature. Significantly, a tri-activity volcano plot was constructed based on the unified activity descriptor of O adsorption energy for guiding the design of SACs in H<sub>2</sub>O<sub>2</sub> catalytic oxidation of NO. The pathway of catalytic oxidation NO by OOH radicals is the dominant route in the system of SACs for the catalytic oxidation of NO by H<sub>2</sub>O<sub>2</sub>. The analysis of Bader charge and electronegativity revealed a linear correlation between the structural properties of the catalysts and the catalytic activity descriptor, which can be used to quickly predict the reactivity of SACs with different coordination environments. This work provides a new pathway for the current NO oxidation and guides future work on catalyst screening and experimental preparation.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"29 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-11-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.4c06149\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c06149","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
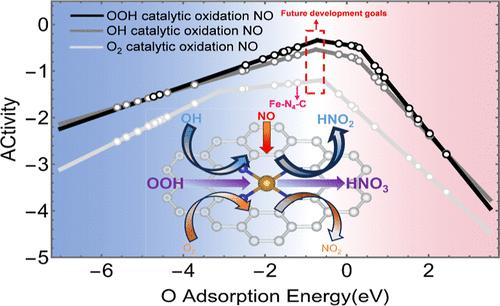
摘要
最近出现的单原子催化剂(SAC)被认为是燃煤发电站氮氧化物减排领域中氮氧化物催化氧化的理想候选催化剂。然而,由于缺乏理论模型的指导,在室温下实现 NO 的高效催化氧化仍是一个主要问题。因此,本研究基于密度泛函理论计算,研究了 OOH 自由基在八种 TM-N4-C(TM = Sc、Cr、Mn、Fe、Co、Ni、Cu 和 Zn)上催化氧化 NO 的反应途径。基于布朗斯特-埃文斯-波兰尼线性关系和微动力学模拟,成功建立并验证了 OOH 催化氧化 NO 的活性火山图模型。在这些催化剂中,Fe-N4-C 和 Mn-N4-C 催化剂的反应活性较高。这两种催化剂决定速率步骤的能量势垒分别为 0.14 和 0.30 eV,说明 NO 催化氧化在室温下是可行的。值得注意的是,根据 O 吸附能这一统一的活性描述符,构建了三活性火山图,用于指导 H2O2 催化氧化 NO 的 SAC 的设计。在 H2O2 催化氧化 NO 的 SACs 体系中,OOH 自由基催化氧化 NO 的途径是主要途径。通过对巴德尔电荷和电负性的分析,发现催化剂的结构特性与催化活性描述符之间存在线性相关,可用于快速预测具有不同配位环境的 SAC 的反应活性。这项工作为当前的氮氧化物氧化提供了一条新途径,并为今后的催化剂筛选和实验制备工作提供了指导。
Probing into the Catalytic Activity of Single-Atom Catalysts for NO Oxidation by H2O2 via the Tri-activity Volcano Plot
The recently emerging single-atom catalysts (SACs) are proposed as promising candidates for the catalytic oxidation of NO in the field of NO pollutant reduction from coal-fired power stations. However, due to the lack of theoretical model guidance, achieving efficient catalytic oxidation of NO at room temperature still remains a major concern. Thus, in this study, the reaction pathways for the catalytic oxidation of NO by OOH radicals on eight TM–N4–C (TM = Sc, Cr, Mn, Fe, Co, Ni, Cu, and Zn) were investigated based on density functional theory calculations. Based on the Bronsted–Evans–Polanyi linear relationship and microkinetic simulations, the activity volcano plot model for the catalytic oxidation of NO by OOH has been successfully established and validated. The Fe–N4–C and Mn–N4–C catalysts showed higher reactivities among these catalysts. The energy barriers for the rate-determining steps of these two catalysts were 0.14 and 0.30 eV, respectively, illustrating that catalytic oxidation of NO is feasible at room temperature. Significantly, a tri-activity volcano plot was constructed based on the unified activity descriptor of O adsorption energy for guiding the design of SACs in H2O2 catalytic oxidation of NO. The pathway of catalytic oxidation NO by OOH radicals is the dominant route in the system of SACs for the catalytic oxidation of NO by H2O2. The analysis of Bader charge and electronegativity revealed a linear correlation between the structural properties of the catalysts and the catalytic activity descriptor, which can be used to quickly predict the reactivity of SACs with different coordination environments. This work provides a new pathway for the current NO oxidation and guides future work on catalyst screening and experimental preparation.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
