Lucas P. Ramos, Felipe A. Louza, Guilherme P. Telles
{"title":"利用简洁彩色德布鲁因图进行比较基因组学研究","authors":"Lucas P. Ramos, Felipe A. Louza, Guilherme P. Telles","doi":"10.1007/s00236-024-00467-7","DOIUrl":null,"url":null,"abstract":"<div><p>DNA technologies have evolved significantly in the past years enabling the sequencing of a large number of genomes in a short time. Nevertheless, the underlying problem of assembling sequence fragments is computationally hard and many technical factors and limitations complicate obtaining the complete sequence of a genome. Many genomes are left in a draft state, in which each chromosome is represented by a set of sequences with partial information on their relative order. Recently, some approaches have been proposed to compare draft genomes by comparing paths in de Bruijn graphs, which are constructed by many practical genome assemblers. In this article we describe in more detail a method for comparing genomes represented as succinct colored de Bruijn graphs directly and without resorting to sequence alignments, called <span>\\(\\texttt {gcBB}\\)</span>, that evaluates the entropy and expectation measures based on the Burrows-Wheeler Similarity Distribution. We also introduce an improved version of <span>\\(\\texttt {gcBB}\\)</span>, called <span>\\(\\texttt {multi-gcBB}\\)</span>, that improves the time and space performance considerably through the selection of different data structures. We have compared phylogenies of 12 Drosophila species obtained by other methods to those obtained with <span>\\(\\texttt {gcBB}\\)</span>, achieving promising results.</p></div>","PeriodicalId":7189,"journal":{"name":"Acta Informatica","volume":"62 1","pages":""},"PeriodicalIF":0.5000,"publicationDate":"2024-11-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Comparative genomics with succinct colored de Bruijn graphs\",\"authors\":\"Lucas P. Ramos, Felipe A. Louza, Guilherme P. Telles\",\"doi\":\"10.1007/s00236-024-00467-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>DNA technologies have evolved significantly in the past years enabling the sequencing of a large number of genomes in a short time. Nevertheless, the underlying problem of assembling sequence fragments is computationally hard and many technical factors and limitations complicate obtaining the complete sequence of a genome. Many genomes are left in a draft state, in which each chromosome is represented by a set of sequences with partial information on their relative order. Recently, some approaches have been proposed to compare draft genomes by comparing paths in de Bruijn graphs, which are constructed by many practical genome assemblers. In this article we describe in more detail a method for comparing genomes represented as succinct colored de Bruijn graphs directly and without resorting to sequence alignments, called <span>\\\\(\\\\texttt {gcBB}\\\\)</span>, that evaluates the entropy and expectation measures based on the Burrows-Wheeler Similarity Distribution. We also introduce an improved version of <span>\\\\(\\\\texttt {gcBB}\\\\)</span>, called <span>\\\\(\\\\texttt {multi-gcBB}\\\\)</span>, that improves the time and space performance considerably through the selection of different data structures. We have compared phylogenies of 12 Drosophila species obtained by other methods to those obtained with <span>\\\\(\\\\texttt {gcBB}\\\\)</span>, achieving promising results.</p></div>\",\"PeriodicalId\":7189,\"journal\":{\"name\":\"Acta Informatica\",\"volume\":\"62 1\",\"pages\":\"\"},\"PeriodicalIF\":0.5000,\"publicationDate\":\"2024-11-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Informatica\",\"FirstCategoryId\":\"94\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00236-024-00467-7\",\"RegionNum\":4,\"RegionCategory\":\"计算机科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"COMPUTER SCIENCE, INFORMATION SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Informatica","FirstCategoryId":"94","ListUrlMain":"https://link.springer.com/article/10.1007/s00236-024-00467-7","RegionNum":4,"RegionCategory":"计算机科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"COMPUTER SCIENCE, INFORMATION SYSTEMS","Score":null,"Total":0}
Comparative genomics with succinct colored de Bruijn graphs
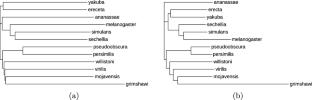
DNA technologies have evolved significantly in the past years enabling the sequencing of a large number of genomes in a short time. Nevertheless, the underlying problem of assembling sequence fragments is computationally hard and many technical factors and limitations complicate obtaining the complete sequence of a genome. Many genomes are left in a draft state, in which each chromosome is represented by a set of sequences with partial information on their relative order. Recently, some approaches have been proposed to compare draft genomes by comparing paths in de Bruijn graphs, which are constructed by many practical genome assemblers. In this article we describe in more detail a method for comparing genomes represented as succinct colored de Bruijn graphs directly and without resorting to sequence alignments, called \(\texttt {gcBB}\), that evaluates the entropy and expectation measures based on the Burrows-Wheeler Similarity Distribution. We also introduce an improved version of \(\texttt {gcBB}\), called \(\texttt {multi-gcBB}\), that improves the time and space performance considerably through the selection of different data structures. We have compared phylogenies of 12 Drosophila species obtained by other methods to those obtained with \(\texttt {gcBB}\), achieving promising results.
期刊介绍:
Acta Informatica provides international dissemination of articles on formal methods for the design and analysis of programs, computing systems and information structures, as well as related fields of Theoretical Computer Science such as Automata Theory, Logic in Computer Science, and Algorithmics.
Topics of interest include:
• semantics of programming languages
• models and modeling languages for concurrent, distributed, reactive and mobile systems
• models and modeling languages for timed, hybrid and probabilistic systems
• specification, program analysis and verification
• model checking and theorem proving
• modal, temporal, first- and higher-order logics, and their variants
• constraint logic, SAT/SMT-solving techniques
• theoretical aspects of databases, semi-structured data and finite model theory
• theoretical aspects of artificial intelligence, knowledge representation, description logic
• automata theory, formal languages, term and graph rewriting
• game-based models, synthesis
• type theory, typed calculi
• algebraic, coalgebraic and categorical methods
• formal aspects of performance, dependability and reliability analysis
• foundations of information and network security
• parallel, distributed and randomized algorithms
• design and analysis of algorithms
• foundations of network and communication protocols.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
