Yang Wang, Zhaofan Li, Andrea Giuntoli* and Wenjie Xia*,
{"title":"通过能量重正化方法实现供体-受体聚合物的热力学和构象一致性粗粒化","authors":"Yang Wang, Zhaofan Li, Andrea Giuntoli* and Wenjie Xia*, ","doi":"10.1021/acsapm.4c03380","DOIUrl":null,"url":null,"abstract":"<p >Understanding the thermomechanical and conformational properties of semiconducting donor–acceptor conjugated polymers (D-A CPs) at a fundamental molecular scale is essential for the design of high-performance devices. However, the substantial computational demands of all-atom (AA) simulations and the complex heterogeneous structures of CPs pose significant challenges in thoroughly investigating the properties of CPs approaching the real devices scale. Herein, leveraging the well-established framework of the energy renormalization (ER) approach, we develop a temperature- and architecture-transferable chemistry-specific coarse-grained (CG) model for poly(diketopyrrolopyrrole-co-thiophene) (PDPPT)-based D-A CPs with significantly improved computational efficiency. Our results show excellent agreement between AA and CG simulations in predicting key properties such as density, Debye–Waller factor, and Young’s modulus across a wide range of temperature and chain architectures. Specifically, the ER-corrected CG model captures trends in glass transition temperature (<i>T</i><sub>g</sub>) and mechanical properties, aligning closely with experimental data. The CG model reveals that longer side chain lengths and less bulky backbone conjugation units induce a lower <i>T</i><sub>g</sub> and Young’s modulus, with bulky backbone units exhibiting slower dynamics. The localization model accurately predicts relaxation times across different molecular architectures. Additionally, the CG model’s conformational properties align with experimental data and theoretical worm-like chain models, showing that persistence length increases with longer side chains, while bulky backbone moieties decrease it. These findings deepen our understanding of the complex interactions between flexible side chains and rigid backbones in CPs with diverse architectures, offering important insights for the strategic design of CPs with tailored properties.</p>","PeriodicalId":7,"journal":{"name":"ACS Applied Polymer Materials","volume":"6 22","pages":"14009–14020 14009–14020"},"PeriodicalIF":4.7000,"publicationDate":"2024-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Thermomechanically and Conformationally Consistent Coarse-Graining of Donor–Acceptor Polymers via Energy Renormalization Approach\",\"authors\":\"Yang Wang, Zhaofan Li, Andrea Giuntoli* and Wenjie Xia*, \",\"doi\":\"10.1021/acsapm.4c03380\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Understanding the thermomechanical and conformational properties of semiconducting donor–acceptor conjugated polymers (D-A CPs) at a fundamental molecular scale is essential for the design of high-performance devices. However, the substantial computational demands of all-atom (AA) simulations and the complex heterogeneous structures of CPs pose significant challenges in thoroughly investigating the properties of CPs approaching the real devices scale. Herein, leveraging the well-established framework of the energy renormalization (ER) approach, we develop a temperature- and architecture-transferable chemistry-specific coarse-grained (CG) model for poly(diketopyrrolopyrrole-co-thiophene) (PDPPT)-based D-A CPs with significantly improved computational efficiency. Our results show excellent agreement between AA and CG simulations in predicting key properties such as density, Debye–Waller factor, and Young’s modulus across a wide range of temperature and chain architectures. Specifically, the ER-corrected CG model captures trends in glass transition temperature (<i>T</i><sub>g</sub>) and mechanical properties, aligning closely with experimental data. The CG model reveals that longer side chain lengths and less bulky backbone conjugation units induce a lower <i>T</i><sub>g</sub> and Young’s modulus, with bulky backbone units exhibiting slower dynamics. The localization model accurately predicts relaxation times across different molecular architectures. Additionally, the CG model’s conformational properties align with experimental data and theoretical worm-like chain models, showing that persistence length increases with longer side chains, while bulky backbone moieties decrease it. These findings deepen our understanding of the complex interactions between flexible side chains and rigid backbones in CPs with diverse architectures, offering important insights for the strategic design of CPs with tailored properties.</p>\",\"PeriodicalId\":7,\"journal\":{\"name\":\"ACS Applied Polymer Materials\",\"volume\":\"6 22\",\"pages\":\"14009–14020 14009–14020\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-11-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Applied Polymer Materials\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsapm.4c03380\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Polymer Materials","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsapm.4c03380","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
Thermomechanically and Conformationally Consistent Coarse-Graining of Donor–Acceptor Polymers via Energy Renormalization Approach
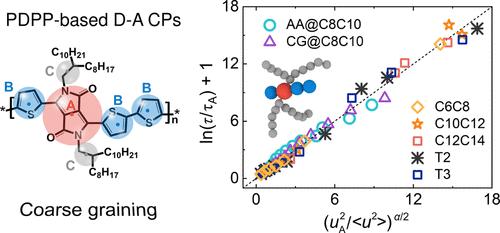
Understanding the thermomechanical and conformational properties of semiconducting donor–acceptor conjugated polymers (D-A CPs) at a fundamental molecular scale is essential for the design of high-performance devices. However, the substantial computational demands of all-atom (AA) simulations and the complex heterogeneous structures of CPs pose significant challenges in thoroughly investigating the properties of CPs approaching the real devices scale. Herein, leveraging the well-established framework of the energy renormalization (ER) approach, we develop a temperature- and architecture-transferable chemistry-specific coarse-grained (CG) model for poly(diketopyrrolopyrrole-co-thiophene) (PDPPT)-based D-A CPs with significantly improved computational efficiency. Our results show excellent agreement between AA and CG simulations in predicting key properties such as density, Debye–Waller factor, and Young’s modulus across a wide range of temperature and chain architectures. Specifically, the ER-corrected CG model captures trends in glass transition temperature (Tg) and mechanical properties, aligning closely with experimental data. The CG model reveals that longer side chain lengths and less bulky backbone conjugation units induce a lower Tg and Young’s modulus, with bulky backbone units exhibiting slower dynamics. The localization model accurately predicts relaxation times across different molecular architectures. Additionally, the CG model’s conformational properties align with experimental data and theoretical worm-like chain models, showing that persistence length increases with longer side chains, while bulky backbone moieties decrease it. These findings deepen our understanding of the complex interactions between flexible side chains and rigid backbones in CPs with diverse architectures, offering important insights for the strategic design of CPs with tailored properties.
期刊介绍:
ACS Applied Polymer Materials is an interdisciplinary journal publishing original research covering all aspects of engineering, chemistry, physics, and biology relevant to applications of polymers.
The journal is devoted to reports of new and original experimental and theoretical research of an applied nature that integrates fundamental knowledge in the areas of materials, engineering, physics, bioscience, polymer science and chemistry into important polymer applications. The journal is specifically interested in work that addresses relationships among structure, processing, morphology, chemistry, properties, and function as well as work that provide insights into mechanisms critical to the performance of the polymer for applications.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
