Guoqiang Ding, Yiwen Gao, Hetong Zhang, Na Yang, Xiaobin Niu and Jianwei Wang
{"title":"掺杂过渡金属原子的 MoSi2N4 单层作为电化学二氧化碳还原反应催化剂的理论研究","authors":"Guoqiang Ding, Yiwen Gao, Hetong Zhang, Na Yang, Xiaobin Niu and Jianwei Wang","doi":"10.1039/D4CP03493G","DOIUrl":null,"url":null,"abstract":"<p >Following the principle of single-atom catalysts (SACs), the fourth-period transition metals (TM) were designed as active sites on a MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> monolayer surface with N vacancy, and the catalytic mechanisms of these single-atom active sites for the conversion of CO<small><sub>2</sub></small> to CO were investigated by first-principles calculations. Our results showed that the doped TM atoms on the MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> surface significantly enhanced the CO<small><sub>2</sub></small> reduction reaction (CO<small><sub>2</sub></small>RR) activity compared with the pristine MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> monolayer. Our findings after analyzing all the doped structures in our work were as follows: (1) the Sc-, Ti-, and Mn-doped structures exhibited very low limiting potentials; (2) out of Sc-, Ti- and Mn-doped structures, the Mn@MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small>-N<small><sub>v</sub></small> structure showed the best catalytic performance with a limiting potential of only –0.16 V, exhibiting an advantage over the hydrogen evolution reaction, which is a competitive reaction of CO<small><sub>2</sub></small>RR. However, the positive binding free energy at 298.15 K of the intermediate reactant *COOH on the Mn@MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small>-N<small><sub>v</sub></small> surface indicated its unstable state, which hinders the CO generation process. This is contrary to the results derived from the adsorption/binding energy at 0 K, which indicated that the effect of temperature cannot be ignored when considering adsorption/binding energy. Our work provides insights into the effects of temperature on the catalytic mechanisms for CO<small><sub>2</sub></small>RR through TM-doped MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> monolayers.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 1","pages":" 577-588"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Theoretical investigations of transition metal atom-doped MoSi2N4 monolayers as catalysts for electrochemical CO2 reduction reactions†\",\"authors\":\"Guoqiang Ding, Yiwen Gao, Hetong Zhang, Na Yang, Xiaobin Niu and Jianwei Wang\",\"doi\":\"10.1039/D4CP03493G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Following the principle of single-atom catalysts (SACs), the fourth-period transition metals (TM) were designed as active sites on a MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> monolayer surface with N vacancy, and the catalytic mechanisms of these single-atom active sites for the conversion of CO<small><sub>2</sub></small> to CO were investigated by first-principles calculations. Our results showed that the doped TM atoms on the MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> surface significantly enhanced the CO<small><sub>2</sub></small> reduction reaction (CO<small><sub>2</sub></small>RR) activity compared with the pristine MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> monolayer. Our findings after analyzing all the doped structures in our work were as follows: (1) the Sc-, Ti-, and Mn-doped structures exhibited very low limiting potentials; (2) out of Sc-, Ti- and Mn-doped structures, the Mn@MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small>-N<small><sub>v</sub></small> structure showed the best catalytic performance with a limiting potential of only –0.16 V, exhibiting an advantage over the hydrogen evolution reaction, which is a competitive reaction of CO<small><sub>2</sub></small>RR. However, the positive binding free energy at 298.15 K of the intermediate reactant *COOH on the Mn@MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small>-N<small><sub>v</sub></small> surface indicated its unstable state, which hinders the CO generation process. This is contrary to the results derived from the adsorption/binding energy at 0 K, which indicated that the effect of temperature cannot be ignored when considering adsorption/binding energy. Our work provides insights into the effects of temperature on the catalytic mechanisms for CO<small><sub>2</sub></small>RR through TM-doped MoSi<small><sub>2</sub></small>N<small><sub>4</sub></small> monolayers.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 1\",\"pages\":\" 577-588\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-11-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03493g\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03493g","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
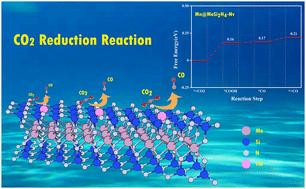
根据单原子催化剂(SAC)的原理,我们在带有 N 空位的 MoSi2N4 单层表面设计了第四周期过渡金属(TM)作为活性位点,并通过第一性原理计算研究了这些单原子活性位点将 CO2 转化为 CO 的催化机理。结果表明,与原始的 MoSi2N4 单层相比,MoSi2N4 表面掺杂的 TM 原子大大提高了 CO2 还原反应(CO2RR)的活性。在我们分析的所有掺杂结构中,我们发现:(1) Sc、Ti 和 Mn 掺杂结构的极限电位非常低;(2) 在 Sc、Ti 和 Mn 掺杂结构中,Mn@MoSi2N4-Nv 结构的催化性能最好,极限电位仅为 -0.16 V,而且在 CO2RR 的竞争反应--氢进化反应中也具有优势。然而,中间反应物 *COOH 在 Mn@MoSi2N4-Nv 表面 298.15 K 时的结合自由能为正值,表明其处于不稳定状态,给 CO 产物的生成过程带来了麻烦。这与 0 K 时的吸附/结合能得出的结果不同,表明在考虑吸附/结合能时不能忽视温度效应。我们的工作为通过掺杂 TM 的 MoSi2N4 单层催化 CO2RR 的机制中的温度效应提供了见解。
Theoretical investigations of transition metal atom-doped MoSi2N4 monolayers as catalysts for electrochemical CO2 reduction reactions†
Following the principle of single-atom catalysts (SACs), the fourth-period transition metals (TM) were designed as active sites on a MoSi2N4 monolayer surface with N vacancy, and the catalytic mechanisms of these single-atom active sites for the conversion of CO2 to CO were investigated by first-principles calculations. Our results showed that the doped TM atoms on the MoSi2N4 surface significantly enhanced the CO2 reduction reaction (CO2RR) activity compared with the pristine MoSi2N4 monolayer. Our findings after analyzing all the doped structures in our work were as follows: (1) the Sc-, Ti-, and Mn-doped structures exhibited very low limiting potentials; (2) out of Sc-, Ti- and Mn-doped structures, the Mn@MoSi2N4-Nv structure showed the best catalytic performance with a limiting potential of only –0.16 V, exhibiting an advantage over the hydrogen evolution reaction, which is a competitive reaction of CO2RR. However, the positive binding free energy at 298.15 K of the intermediate reactant *COOH on the Mn@MoSi2N4-Nv surface indicated its unstable state, which hinders the CO generation process. This is contrary to the results derived from the adsorption/binding energy at 0 K, which indicated that the effect of temperature cannot be ignored when considering adsorption/binding energy. Our work provides insights into the effects of temperature on the catalytic mechanisms for CO2RR through TM-doped MoSi2N4 monolayers.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
