{"title":"格里斯切利综合征 2 型:对 149 例新发现和既往描述的 RAB27A 缺乏症患者的综合分析。","authors":"Jesmeen Maimaris, Adriel Roa-Bautista, Mahreen Sohail, Claire Booth, Chiara Cugno, Lenka Chenchara, Tawfeg Ben Omran, Yael Hacohen, Ming Lim, Kimberly Gilmour, Gillian Griffiths, Kanchan Rao, Reem Elfeky, Maaike Kusters","doi":"10.1007/s10875-024-01842-2","DOIUrl":null,"url":null,"abstract":"<p><p>Griscelli syndrome type 2 (GS2) is a rare, life-threatening immunodysregulatory disorder characterised by impaired cytotoxic activity leading to susceptibility to haemophagocytic lymphohistiocytosis (HLH) and hypopigmentation. We completed a literature review and analysis of clinical data of 149 patients with GS2 including 8 new patients.We identified three founder mutations which show diverse phenotypic profiles (RAB27A c.244 C > T, p.R82C, c.514_518delCAAGC, p.Q172NfsX2, c.550 C > T, p.R184X). The most common presentation was HLH (119/149, 80%), with high proportion of central nervous system involvement (68/149, 46%). Features of partial albinism were present in 105 of 149 cases (70%). Hypopigmentation can be absent in GS2 and should not exclude the diagnosis. Patients with biallelic protein truncating variants (PTV) were more likely to have systemic HLH (44/56, 79%) and partial albinism (45/56, 80%), in comparison to hypomorphic variants (9/41, 22%; 20/41, 49%). Patients with hypomorphic variants presented later (5.4 years cf. 0.4 years, p = < 0.0001) and were more likely to have isolated CNS HLH (2% cf. 42%, p = 0.001).Mortality was high in the cohort (50/149, 34%). Survival of cases post-HLH who underwent transplantation is superior to un-transplanted patients, suggesting adequate HLH control followed by early HSCT is highly beneficial. Mortality was reduced in HSCT recipients versus the un-transplanted group where follow-up data was available (14% compared to 58%).Asymptomatic cases identified through family history/genetic screening may benefit from pre-emptive HSCT, but access and development of robust functional testing are required. High mortality related to HLH remains concerning and emphasises the need for improved molecular characterisation and clinical prognostic factors to guide management decisions.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"50"},"PeriodicalIF":4.1000,"publicationDate":"2024-11-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11604824/pdf/","citationCount":"0","resultStr":"{\"title\":\"Griscelli Syndrome Type 2: Comprehensive Analysis of 149 New and Previously Described Patients with RAB27A Deficiency.\",\"authors\":\"Jesmeen Maimaris, Adriel Roa-Bautista, Mahreen Sohail, Claire Booth, Chiara Cugno, Lenka Chenchara, Tawfeg Ben Omran, Yael Hacohen, Ming Lim, Kimberly Gilmour, Gillian Griffiths, Kanchan Rao, Reem Elfeky, Maaike Kusters\",\"doi\":\"10.1007/s10875-024-01842-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Griscelli syndrome type 2 (GS2) is a rare, life-threatening immunodysregulatory disorder characterised by impaired cytotoxic activity leading to susceptibility to haemophagocytic lymphohistiocytosis (HLH) and hypopigmentation. We completed a literature review and analysis of clinical data of 149 patients with GS2 including 8 new patients.We identified three founder mutations which show diverse phenotypic profiles (RAB27A c.244 C > T, p.R82C, c.514_518delCAAGC, p.Q172NfsX2, c.550 C > T, p.R184X). The most common presentation was HLH (119/149, 80%), with high proportion of central nervous system involvement (68/149, 46%). Features of partial albinism were present in 105 of 149 cases (70%). Hypopigmentation can be absent in GS2 and should not exclude the diagnosis. Patients with biallelic protein truncating variants (PTV) were more likely to have systemic HLH (44/56, 79%) and partial albinism (45/56, 80%), in comparison to hypomorphic variants (9/41, 22%; 20/41, 49%). Patients with hypomorphic variants presented later (5.4 years cf. 0.4 years, p = < 0.0001) and were more likely to have isolated CNS HLH (2% cf. 42%, p = 0.001).Mortality was high in the cohort (50/149, 34%). Survival of cases post-HLH who underwent transplantation is superior to un-transplanted patients, suggesting adequate HLH control followed by early HSCT is highly beneficial. Mortality was reduced in HSCT recipients versus the un-transplanted group where follow-up data was available (14% compared to 58%).Asymptomatic cases identified through family history/genetic screening may benefit from pre-emptive HSCT, but access and development of robust functional testing are required. High mortality related to HLH remains concerning and emphasises the need for improved molecular characterisation and clinical prognostic factors to guide management decisions.</p>\",\"PeriodicalId\":15531,\"journal\":{\"name\":\"Journal of Clinical Immunology\",\"volume\":\"45 1\",\"pages\":\"50\"},\"PeriodicalIF\":4.1000,\"publicationDate\":\"2024-11-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11604824/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10875-024-01842-2\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01842-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Griscelli Syndrome Type 2: Comprehensive Analysis of 149 New and Previously Described Patients with RAB27A Deficiency.
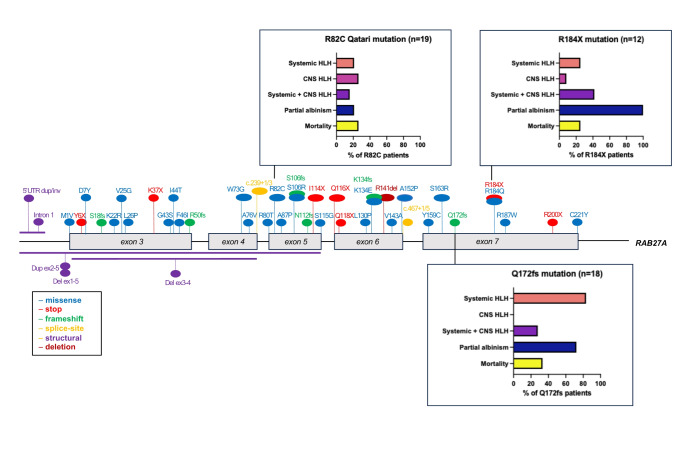
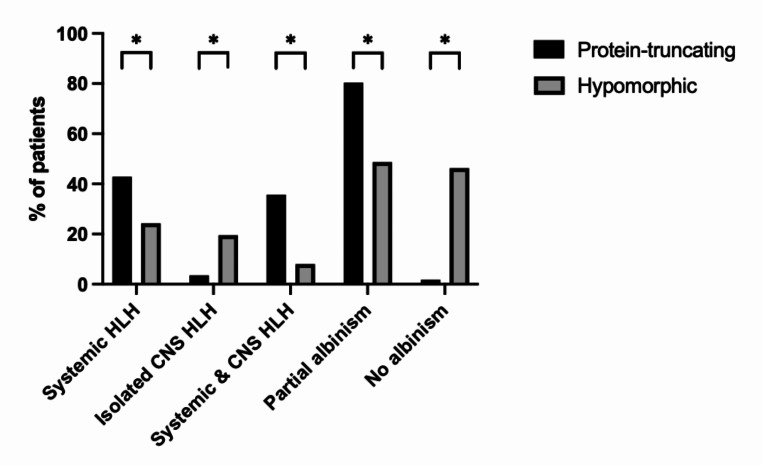
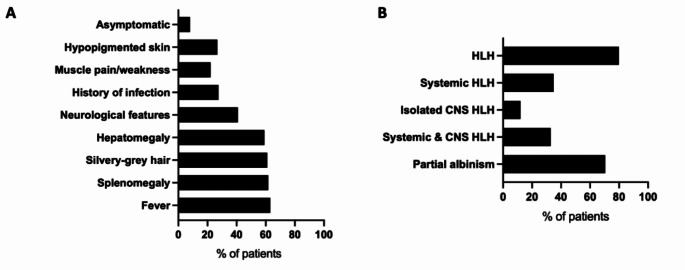
Griscelli syndrome type 2 (GS2) is a rare, life-threatening immunodysregulatory disorder characterised by impaired cytotoxic activity leading to susceptibility to haemophagocytic lymphohistiocytosis (HLH) and hypopigmentation. We completed a literature review and analysis of clinical data of 149 patients with GS2 including 8 new patients.We identified three founder mutations which show diverse phenotypic profiles (RAB27A c.244 C > T, p.R82C, c.514_518delCAAGC, p.Q172NfsX2, c.550 C > T, p.R184X). The most common presentation was HLH (119/149, 80%), with high proportion of central nervous system involvement (68/149, 46%). Features of partial albinism were present in 105 of 149 cases (70%). Hypopigmentation can be absent in GS2 and should not exclude the diagnosis. Patients with biallelic protein truncating variants (PTV) were more likely to have systemic HLH (44/56, 79%) and partial albinism (45/56, 80%), in comparison to hypomorphic variants (9/41, 22%; 20/41, 49%). Patients with hypomorphic variants presented later (5.4 years cf. 0.4 years, p = < 0.0001) and were more likely to have isolated CNS HLH (2% cf. 42%, p = 0.001).Mortality was high in the cohort (50/149, 34%). Survival of cases post-HLH who underwent transplantation is superior to un-transplanted patients, suggesting adequate HLH control followed by early HSCT is highly beneficial. Mortality was reduced in HSCT recipients versus the un-transplanted group where follow-up data was available (14% compared to 58%).Asymptomatic cases identified through family history/genetic screening may benefit from pre-emptive HSCT, but access and development of robust functional testing are required. High mortality related to HLH remains concerning and emphasises the need for improved molecular characterisation and clinical prognostic factors to guide management decisions.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
