Amy Marie Campbell, Chris Hauton, Ronny van Aerle, Jaime Martinez-Urtaza
{"title":"副溶血性弧菌序列3型扩展的生态进化驱动因素:回顾性机器学习方法。","authors":"Amy Marie Campbell, Chris Hauton, Ronny van Aerle, Jaime Martinez-Urtaza","doi":"10.2196/62747","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Environmentally sensitive pathogens exhibit ecological and evolutionary responses to climate change that result in the emergence and global expansion of well-adapted variants. It is imperative to understand the mechanisms that facilitate pathogen emergence and expansion, as well as the drivers behind the mechanisms, to understand and prepare for future pandemic expansions.</p><p><strong>Objective: </strong>The unique, rapid, global expansion of a clonal complex of Vibrio parahaemolyticus (a marine bacterium causing gastroenteritis infections) named Vibrio parahaemolyticus sequence type 3 (VpST3) provides an opportunity to explore the eco-evolutionary drivers of pathogen expansion.</p><p><strong>Methods: </strong>The global expansion of VpST3 was reconstructed using VpST3 genomes, which were then classified into metrics characterizing the stages of this expansion process, indicative of the stages of emergence and establishment. We used machine learning, specifically a random forest classifier, to test a range of ecological and evolutionary drivers for their potential in predicting VpST3 expansion dynamics.</p><p><strong>Results: </strong>We identified a range of evolutionary features, including mutations in the core genome and accessory gene presence, associated with expansion dynamics. A range of random forest classifier approaches were tested to predict expansion classification metrics for each genome. The highest predictive accuracies (ranging from 0.722 to 0.967) were achieved for models using a combined eco-evolutionary approach. While population structure and the difference between introduced and established isolates could be predicted to a high accuracy, our model reported multiple false positives when predicting the success of an introduced isolate, suggesting potential limiting factors not represented in our eco-evolutionary features. Regional models produced for 2 countries reporting the most VpST3 genomes had varying success, reflecting the impacts of class imbalance.</p><p><strong>Conclusions: </strong>These novel insights into evolutionary features and ecological conditions related to the stages of VpST3 expansion showcase the potential of machine learning models using genomic data and will contribute to the future understanding of the eco-evolutionary pathways of climate-sensitive pathogens.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":"5 ","pages":"e62747"},"PeriodicalIF":0.0000,"publicationDate":"2024-11-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11638695/pdf/","citationCount":"0","resultStr":"{\"title\":\"Eco-Evolutionary Drivers of Vibrio parahaemolyticus Sequence Type 3 Expansion: Retrospective Machine Learning Approach.\",\"authors\":\"Amy Marie Campbell, Chris Hauton, Ronny van Aerle, Jaime Martinez-Urtaza\",\"doi\":\"10.2196/62747\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Environmentally sensitive pathogens exhibit ecological and evolutionary responses to climate change that result in the emergence and global expansion of well-adapted variants. It is imperative to understand the mechanisms that facilitate pathogen emergence and expansion, as well as the drivers behind the mechanisms, to understand and prepare for future pandemic expansions.</p><p><strong>Objective: </strong>The unique, rapid, global expansion of a clonal complex of Vibrio parahaemolyticus (a marine bacterium causing gastroenteritis infections) named Vibrio parahaemolyticus sequence type 3 (VpST3) provides an opportunity to explore the eco-evolutionary drivers of pathogen expansion.</p><p><strong>Methods: </strong>The global expansion of VpST3 was reconstructed using VpST3 genomes, which were then classified into metrics characterizing the stages of this expansion process, indicative of the stages of emergence and establishment. We used machine learning, specifically a random forest classifier, to test a range of ecological and evolutionary drivers for their potential in predicting VpST3 expansion dynamics.</p><p><strong>Results: </strong>We identified a range of evolutionary features, including mutations in the core genome and accessory gene presence, associated with expansion dynamics. A range of random forest classifier approaches were tested to predict expansion classification metrics for each genome. The highest predictive accuracies (ranging from 0.722 to 0.967) were achieved for models using a combined eco-evolutionary approach. While population structure and the difference between introduced and established isolates could be predicted to a high accuracy, our model reported multiple false positives when predicting the success of an introduced isolate, suggesting potential limiting factors not represented in our eco-evolutionary features. Regional models produced for 2 countries reporting the most VpST3 genomes had varying success, reflecting the impacts of class imbalance.</p><p><strong>Conclusions: </strong>These novel insights into evolutionary features and ecological conditions related to the stages of VpST3 expansion showcase the potential of machine learning models using genomic data and will contribute to the future understanding of the eco-evolutionary pathways of climate-sensitive pathogens.</p>\",\"PeriodicalId\":73552,\"journal\":{\"name\":\"JMIR bioinformatics and biotechnology\",\"volume\":\"5 \",\"pages\":\"e62747\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-11-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11638695/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JMIR bioinformatics and biotechnology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2196/62747\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/62747","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
背景:环境敏感病原体对气候变化表现出生态和进化反应,导致适应良好的变异的出现和全球扩张。必须了解促进病原体出现和扩展的机制,以及这些机制背后的驱动因素,以便了解和为未来的大流行扩展做好准备。目的:副溶血性弧菌(一种引起胃肠炎感染的海洋细菌)克隆复合体VpST3 (Vibrio parahaolyticus sequence type 3)的独特、快速、全球扩展为探索病原体扩展的生态进化驱动因素提供了机会。方法:利用VpST3基因组重建VpST3的全球扩展,然后将其分类为表征该扩展过程阶段的指标,指示其出现和建立阶段。我们使用机器学习,特别是随机森林分类器,来测试一系列生态和进化驱动因素在预测VpST3扩展动态方面的潜力。结果:我们发现了一系列进化特征,包括核心基因组的突变和辅助基因的存在,这些特征与扩张动力学有关。测试了一系列随机森林分类器方法来预测每个基因组的扩展分类指标。采用综合生态进化方法的模型预测精度最高,为0.722 ~ 0.967。虽然种群结构和引入菌株和已建立菌株之间的差异可以预测到很高的准确性,但我们的模型在预测引入菌株的成功时报告了多个假阳性,这表明潜在的限制因素没有在我们的生态进化特征中得到体现。为报告VpST3基因组最多的两个国家制作的区域模型取得了不同程度的成功,反映了阶级不平衡的影响。结论:这些关于VpST3扩展阶段相关的进化特征和生态条件的新见解展示了使用基因组数据的机器学习模型的潜力,并将有助于未来了解气候敏感病原体的生态进化途径。
Eco-Evolutionary Drivers of Vibrio parahaemolyticus Sequence Type 3 Expansion: Retrospective Machine Learning Approach.
Background: Environmentally sensitive pathogens exhibit ecological and evolutionary responses to climate change that result in the emergence and global expansion of well-adapted variants. It is imperative to understand the mechanisms that facilitate pathogen emergence and expansion, as well as the drivers behind the mechanisms, to understand and prepare for future pandemic expansions.
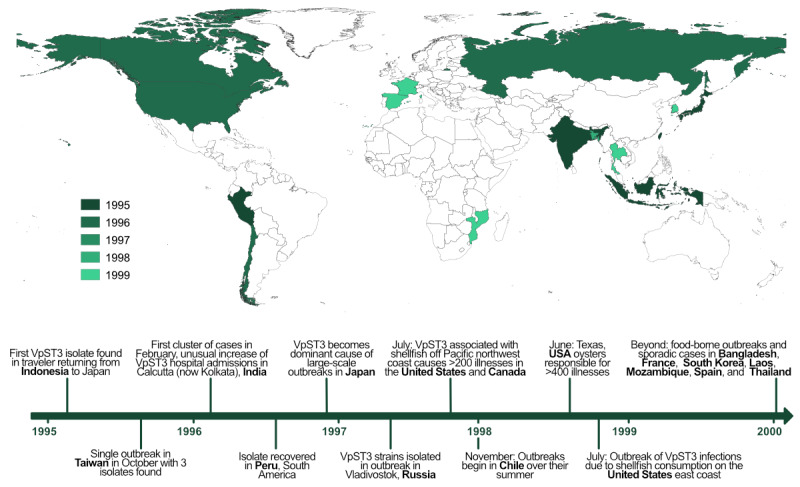
Objective: The unique, rapid, global expansion of a clonal complex of Vibrio parahaemolyticus (a marine bacterium causing gastroenteritis infections) named Vibrio parahaemolyticus sequence type 3 (VpST3) provides an opportunity to explore the eco-evolutionary drivers of pathogen expansion.
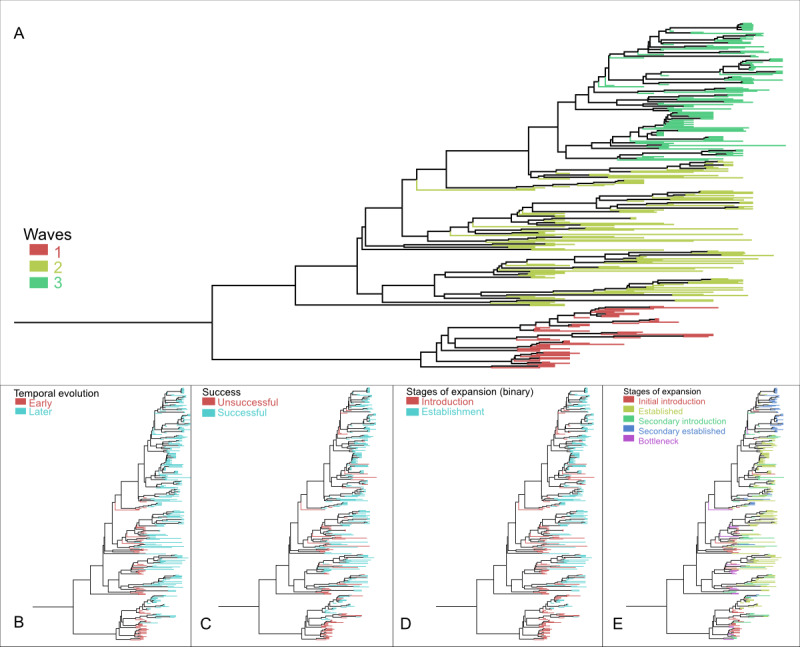
Methods: The global expansion of VpST3 was reconstructed using VpST3 genomes, which were then classified into metrics characterizing the stages of this expansion process, indicative of the stages of emergence and establishment. We used machine learning, specifically a random forest classifier, to test a range of ecological and evolutionary drivers for their potential in predicting VpST3 expansion dynamics.
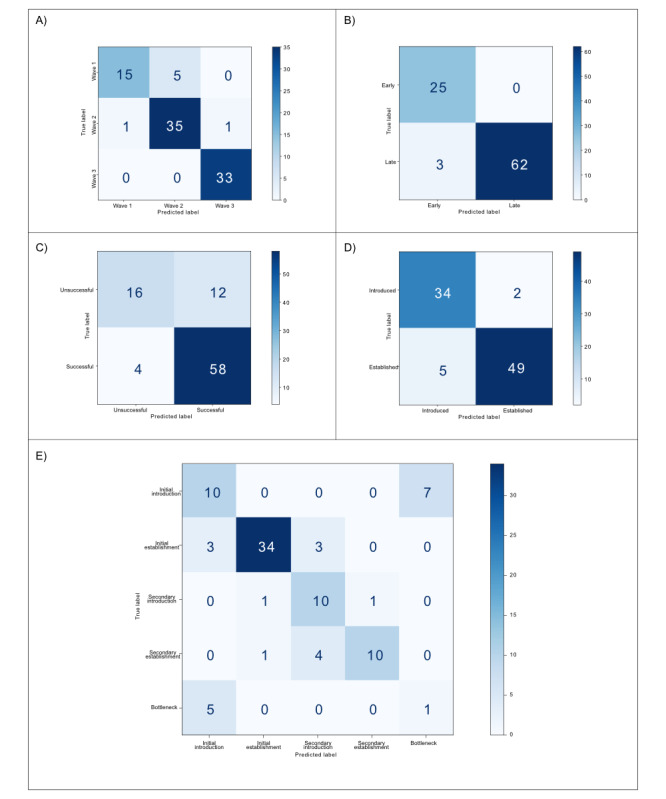
Results: We identified a range of evolutionary features, including mutations in the core genome and accessory gene presence, associated with expansion dynamics. A range of random forest classifier approaches were tested to predict expansion classification metrics for each genome. The highest predictive accuracies (ranging from 0.722 to 0.967) were achieved for models using a combined eco-evolutionary approach. While population structure and the difference between introduced and established isolates could be predicted to a high accuracy, our model reported multiple false positives when predicting the success of an introduced isolate, suggesting potential limiting factors not represented in our eco-evolutionary features. Regional models produced for 2 countries reporting the most VpST3 genomes had varying success, reflecting the impacts of class imbalance.
Conclusions: These novel insights into evolutionary features and ecological conditions related to the stages of VpST3 expansion showcase the potential of machine learning models using genomic data and will contribute to the future understanding of the eco-evolutionary pathways of climate-sensitive pathogens.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
