Xin Zhen, Michael J Betti, Meltem Ece Kars, Andrew R Patterson, Edgar Alejandro Medina-Torres, Selma Cecilia Scheffler Mendoza, Diana Andrea Herrera Sánchez, Gabriela Lopez-Herrera, Yevgeniya Svyryd, Osvaldo M Mutchinick, Eric R Gamazon, Jeffrey C Rathmell, Yuval Itan, Janet Markle, Patricia O'Farrill Romanillos, Saul Oswaldo Lugo-Reyes, Ruben Martinez-Barricarte
{"title":"一种导致G6PC3缺乏的方正突变的分子和临床特征","authors":"Xin Zhen, Michael J Betti, Meltem Ece Kars, Andrew R Patterson, Edgar Alejandro Medina-Torres, Selma Cecilia Scheffler Mendoza, Diana Andrea Herrera Sánchez, Gabriela Lopez-Herrera, Yevgeniya Svyryd, Osvaldo M Mutchinick, Eric R Gamazon, Jeffrey C Rathmell, Yuval Itan, Janet Markle, Patricia O'Farrill Romanillos, Saul Oswaldo Lugo-Reyes, Ruben Martinez-Barricarte","doi":"10.1007/s10875-024-01836-0","DOIUrl":null,"url":null,"abstract":"<p><p>G6PC3 deficiency is a monogenic immunometabolic disorder that causes severe congenital neutropenia type 4. Patients display heterogeneous extra-hematological manifestations, contributing to delayed diagnosis. Here, we investigated the origin and functional consequence of the G6PC3 c.210delC variant found in patients of Mexican descent. Based on the shared haplotypes amongst mutation carriers, we estimated that this variant originated from a founder effect in a common ancestor. Furthermore, by ancestry analysis, we concluded that it appeared in the indigenous Mexican population. At the protein level, we showed that this frameshift mutation leads to an aberrant protein expression in overexpression and patient-derived Epstein-Barr Virus-immortalized B (EBV-B) cells. The neutropenia observed in G6PC3-deficient patients is driven by the intracellular accumulation of the metabolite 1,5-anhydroglucitol-6-phosphate (1,5-AG6P) that inhibits glycolysis. We characterized how the c.210delC variant impacts glycolysis by performing extracellular flux assays on patient-derived EBV-B cells. When treated with 1,5-anhydroglucitol (1,5-AG), the precursor to 1,5-AG6P, patient cells exhibited markedly reduced engagement of glycolysis. Finally, we compared the clinical presentation of patients with the mutation c.210delC and all other G6PC3-deficient patients reported in the literature, and we found that the c.210delC carriers display all prominent clinical features observed in prior patients. In conclusion, G6PC3 c.210delC is a loss-of-function mutation that arose from a founder effect in the indigenous Mexican population. These findings may facilitate the diagnosis of additional patients in this geographical area. Moreover, the in vitro 1,5-AG-dependent functional assay used in our study could be employed to assess the pathogenicity of additional G6PC3 variants.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"53"},"PeriodicalIF":5.7000,"publicationDate":"2024-12-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11618172/pdf/","citationCount":"0","resultStr":"{\"title\":\"Molecular and Clinical Characterization of a Founder Mutation Causing G6PC3 Deficiency.\",\"authors\":\"Xin Zhen, Michael J Betti, Meltem Ece Kars, Andrew R Patterson, Edgar Alejandro Medina-Torres, Selma Cecilia Scheffler Mendoza, Diana Andrea Herrera Sánchez, Gabriela Lopez-Herrera, Yevgeniya Svyryd, Osvaldo M Mutchinick, Eric R Gamazon, Jeffrey C Rathmell, Yuval Itan, Janet Markle, Patricia O'Farrill Romanillos, Saul Oswaldo Lugo-Reyes, Ruben Martinez-Barricarte\",\"doi\":\"10.1007/s10875-024-01836-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>G6PC3 deficiency is a monogenic immunometabolic disorder that causes severe congenital neutropenia type 4. Patients display heterogeneous extra-hematological manifestations, contributing to delayed diagnosis. Here, we investigated the origin and functional consequence of the G6PC3 c.210delC variant found in patients of Mexican descent. Based on the shared haplotypes amongst mutation carriers, we estimated that this variant originated from a founder effect in a common ancestor. Furthermore, by ancestry analysis, we concluded that it appeared in the indigenous Mexican population. At the protein level, we showed that this frameshift mutation leads to an aberrant protein expression in overexpression and patient-derived Epstein-Barr Virus-immortalized B (EBV-B) cells. The neutropenia observed in G6PC3-deficient patients is driven by the intracellular accumulation of the metabolite 1,5-anhydroglucitol-6-phosphate (1,5-AG6P) that inhibits glycolysis. We characterized how the c.210delC variant impacts glycolysis by performing extracellular flux assays on patient-derived EBV-B cells. When treated with 1,5-anhydroglucitol (1,5-AG), the precursor to 1,5-AG6P, patient cells exhibited markedly reduced engagement of glycolysis. Finally, we compared the clinical presentation of patients with the mutation c.210delC and all other G6PC3-deficient patients reported in the literature, and we found that the c.210delC carriers display all prominent clinical features observed in prior patients. In conclusion, G6PC3 c.210delC is a loss-of-function mutation that arose from a founder effect in the indigenous Mexican population. These findings may facilitate the diagnosis of additional patients in this geographical area. Moreover, the in vitro 1,5-AG-dependent functional assay used in our study could be employed to assess the pathogenicity of additional G6PC3 variants.</p>\",\"PeriodicalId\":15531,\"journal\":{\"name\":\"Journal of Clinical Immunology\",\"volume\":\"45 1\",\"pages\":\"53\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-12-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11618172/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10875-024-01836-0\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01836-0","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Molecular and Clinical Characterization of a Founder Mutation Causing G6PC3 Deficiency.
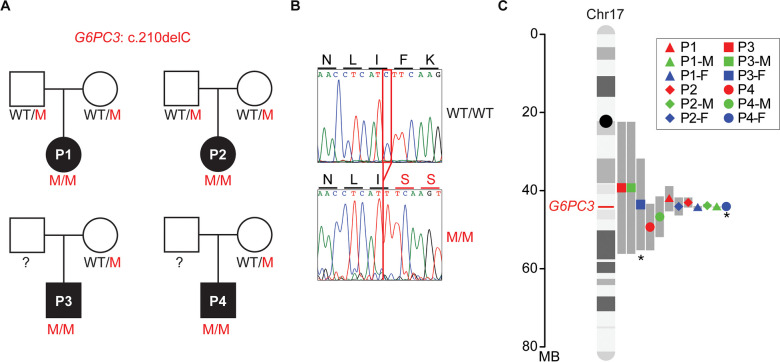
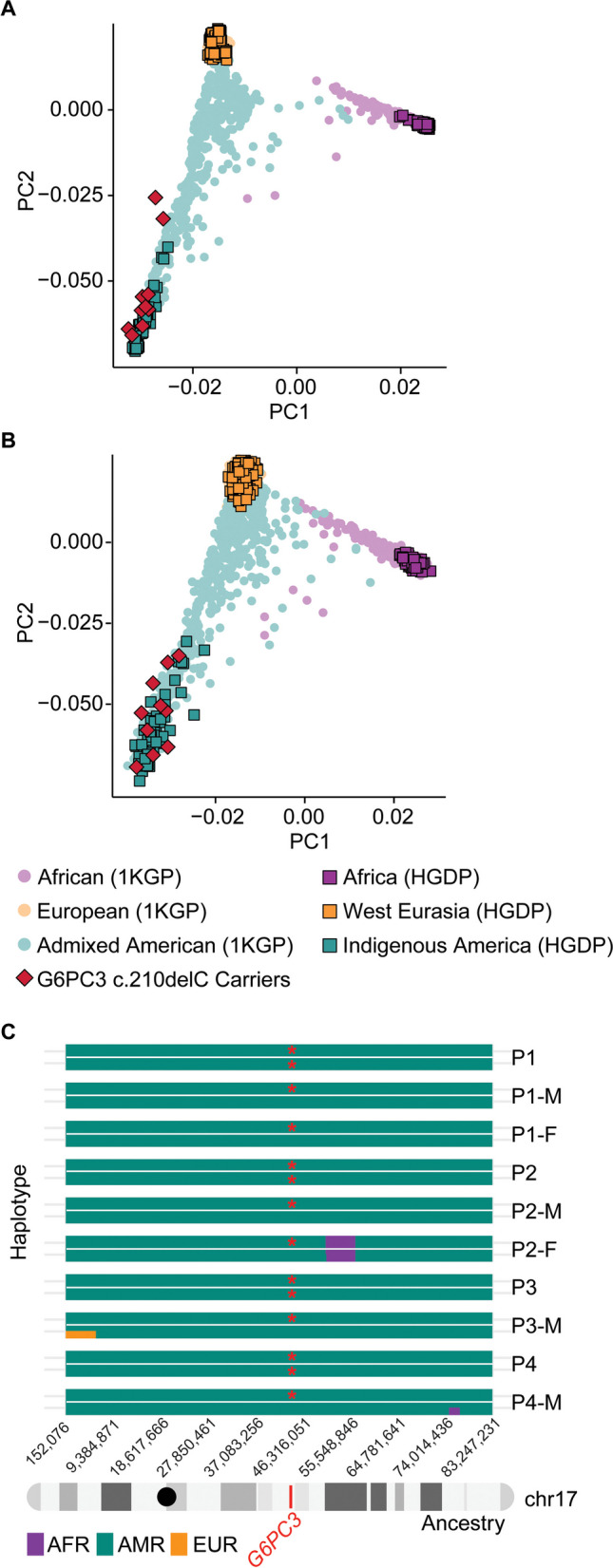
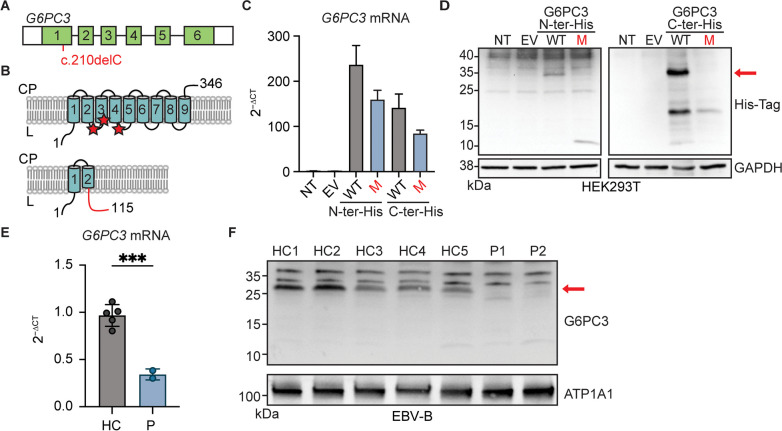
G6PC3 deficiency is a monogenic immunometabolic disorder that causes severe congenital neutropenia type 4. Patients display heterogeneous extra-hematological manifestations, contributing to delayed diagnosis. Here, we investigated the origin and functional consequence of the G6PC3 c.210delC variant found in patients of Mexican descent. Based on the shared haplotypes amongst mutation carriers, we estimated that this variant originated from a founder effect in a common ancestor. Furthermore, by ancestry analysis, we concluded that it appeared in the indigenous Mexican population. At the protein level, we showed that this frameshift mutation leads to an aberrant protein expression in overexpression and patient-derived Epstein-Barr Virus-immortalized B (EBV-B) cells. The neutropenia observed in G6PC3-deficient patients is driven by the intracellular accumulation of the metabolite 1,5-anhydroglucitol-6-phosphate (1,5-AG6P) that inhibits glycolysis. We characterized how the c.210delC variant impacts glycolysis by performing extracellular flux assays on patient-derived EBV-B cells. When treated with 1,5-anhydroglucitol (1,5-AG), the precursor to 1,5-AG6P, patient cells exhibited markedly reduced engagement of glycolysis. Finally, we compared the clinical presentation of patients with the mutation c.210delC and all other G6PC3-deficient patients reported in the literature, and we found that the c.210delC carriers display all prominent clinical features observed in prior patients. In conclusion, G6PC3 c.210delC is a loss-of-function mutation that arose from a founder effect in the indigenous Mexican population. These findings may facilitate the diagnosis of additional patients in this geographical area. Moreover, the in vitro 1,5-AG-dependent functional assay used in our study could be employed to assess the pathogenicity of additional G6PC3 variants.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
