Saige K Power, Sridevi Venkatesan, Sarah Qu, JoAnne McLaurin, Evelyn K Lambe
{"title":"增强的前额叶尼古丁信号作为阿尔茨海默病模型中主动代偿的证据。","authors":"Saige K Power, Sridevi Venkatesan, Sarah Qu, JoAnne McLaurin, Evelyn K Lambe","doi":"10.1186/s40035-024-00452-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cognitive reserve allows for resilience to neuropathology, potentially through active compensation. Here, we examine ex vivo electrophysiological evidence for active compensation in Alzheimer's disease (AD) focusing on the cholinergic innervation of layer 6 in prefrontal cortex. Cholinergic pathways are vulnerable to neuropathology in AD and its preclinical models, and their modulation of deep layer prefrontal cortex is essential for attention and executive function.</p><p><strong>Methods: </strong>We functionally interrogated cholinergic modulation of prefrontal layer 6 pyramidal neurons in two preclinical models: a compound transgenic AD mouse model that permits optogenetically-triggered release of endogenous acetylcholine and a transgenic AD rat model that closely recapitulates the human trajectory of AD. We then tested the impact of therapeutic interventions to further amplify the compensated responses and preserve the typical kinetic profile of cholinergic signaling.</p><p><strong>Results: </strong>In two AD models, we found potentially compensatory upregulation of functional cholinergic responses above non-transgenic controls after onset of pathology. To identify the locus of this enhanced cholinergic signal, we dissected key pre- and post-synaptic components with pharmacological strategies. We identified a significant and selective increase in post-synaptic nicotinic receptor signalling on prefrontal cortical neurons. To probe the additional impact of therapeutic intervention on the adapted circuit, we tested cholinergic and nicotinic-selective pro-cognitive treatments. Inhibition of acetylcholinesterase further enhanced endogenous cholinergic responses but greatly distorted their kinetics. Positive allosteric modulation of nicotinic receptors, by contrast, enhanced endogenous cholinergic responses and retained their rapid kinetics.</p><p><strong>Conclusions: </strong>We demonstrate that functional nicotinic upregulation occurs within the prefrontal cortex in two AD models. Promisingly, this nicotinic signal can be further enhanced while preserving its rapid kinetic signature. Taken together, our work suggests that compensatory mechanisms are active within the prefrontal cortex that can be harnessed by nicotinic receptor positive allosteric modulation, highlighting a new direction for cognitive treatment in AD neuropathology.</p>","PeriodicalId":23269,"journal":{"name":"Translational Neurodegeneration","volume":"13 1","pages":"58"},"PeriodicalIF":15.2000,"publicationDate":"2024-12-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11613856/pdf/","citationCount":"0","resultStr":"{\"title\":\"Enhanced prefrontal nicotinic signaling as evidence of active compensation in Alzheimer's disease models.\",\"authors\":\"Saige K Power, Sridevi Venkatesan, Sarah Qu, JoAnne McLaurin, Evelyn K Lambe\",\"doi\":\"10.1186/s40035-024-00452-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Cognitive reserve allows for resilience to neuropathology, potentially through active compensation. Here, we examine ex vivo electrophysiological evidence for active compensation in Alzheimer's disease (AD) focusing on the cholinergic innervation of layer 6 in prefrontal cortex. Cholinergic pathways are vulnerable to neuropathology in AD and its preclinical models, and their modulation of deep layer prefrontal cortex is essential for attention and executive function.</p><p><strong>Methods: </strong>We functionally interrogated cholinergic modulation of prefrontal layer 6 pyramidal neurons in two preclinical models: a compound transgenic AD mouse model that permits optogenetically-triggered release of endogenous acetylcholine and a transgenic AD rat model that closely recapitulates the human trajectory of AD. We then tested the impact of therapeutic interventions to further amplify the compensated responses and preserve the typical kinetic profile of cholinergic signaling.</p><p><strong>Results: </strong>In two AD models, we found potentially compensatory upregulation of functional cholinergic responses above non-transgenic controls after onset of pathology. To identify the locus of this enhanced cholinergic signal, we dissected key pre- and post-synaptic components with pharmacological strategies. We identified a significant and selective increase in post-synaptic nicotinic receptor signalling on prefrontal cortical neurons. To probe the additional impact of therapeutic intervention on the adapted circuit, we tested cholinergic and nicotinic-selective pro-cognitive treatments. Inhibition of acetylcholinesterase further enhanced endogenous cholinergic responses but greatly distorted their kinetics. Positive allosteric modulation of nicotinic receptors, by contrast, enhanced endogenous cholinergic responses and retained their rapid kinetics.</p><p><strong>Conclusions: </strong>We demonstrate that functional nicotinic upregulation occurs within the prefrontal cortex in two AD models. Promisingly, this nicotinic signal can be further enhanced while preserving its rapid kinetic signature. Taken together, our work suggests that compensatory mechanisms are active within the prefrontal cortex that can be harnessed by nicotinic receptor positive allosteric modulation, highlighting a new direction for cognitive treatment in AD neuropathology.</p>\",\"PeriodicalId\":23269,\"journal\":{\"name\":\"Translational Neurodegeneration\",\"volume\":\"13 1\",\"pages\":\"58\"},\"PeriodicalIF\":15.2000,\"publicationDate\":\"2024-12-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11613856/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational Neurodegeneration\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s40035-024-00452-7\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational Neurodegeneration","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40035-024-00452-7","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
Enhanced prefrontal nicotinic signaling as evidence of active compensation in Alzheimer's disease models.
Background: Cognitive reserve allows for resilience to neuropathology, potentially through active compensation. Here, we examine ex vivo electrophysiological evidence for active compensation in Alzheimer's disease (AD) focusing on the cholinergic innervation of layer 6 in prefrontal cortex. Cholinergic pathways are vulnerable to neuropathology in AD and its preclinical models, and their modulation of deep layer prefrontal cortex is essential for attention and executive function.
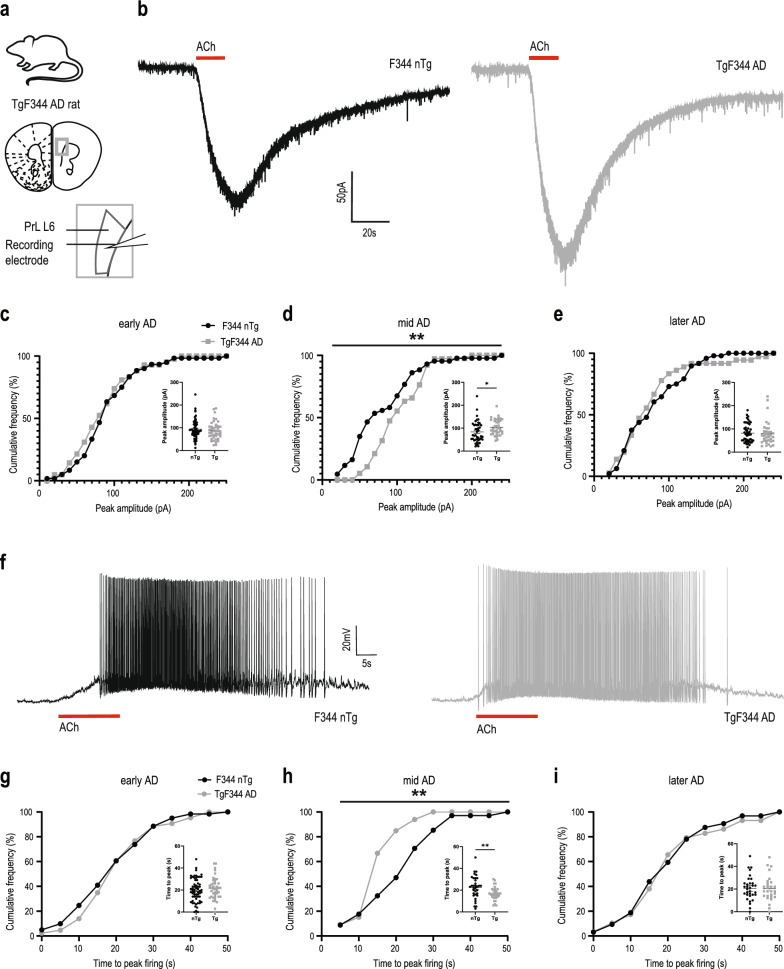
Methods: We functionally interrogated cholinergic modulation of prefrontal layer 6 pyramidal neurons in two preclinical models: a compound transgenic AD mouse model that permits optogenetically-triggered release of endogenous acetylcholine and a transgenic AD rat model that closely recapitulates the human trajectory of AD. We then tested the impact of therapeutic interventions to further amplify the compensated responses and preserve the typical kinetic profile of cholinergic signaling.
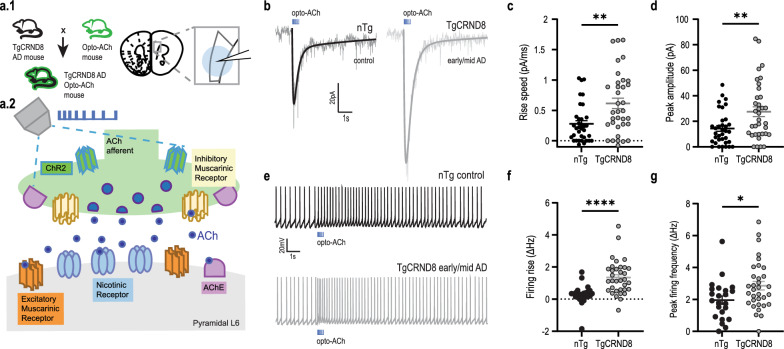
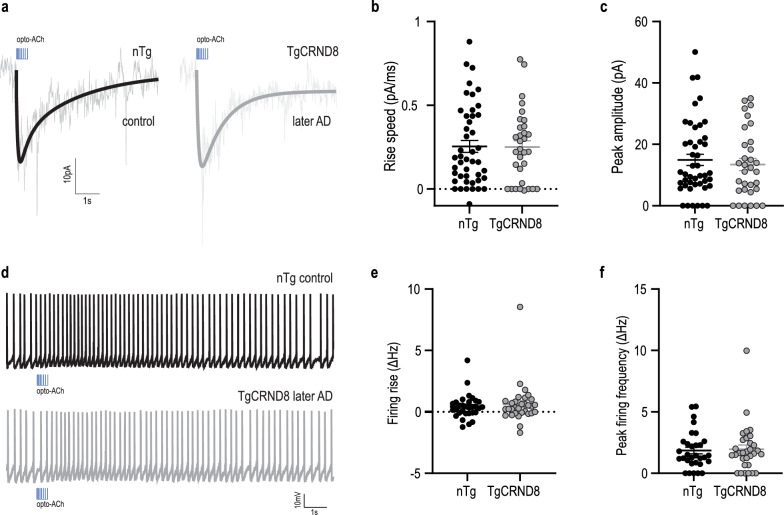
Results: In two AD models, we found potentially compensatory upregulation of functional cholinergic responses above non-transgenic controls after onset of pathology. To identify the locus of this enhanced cholinergic signal, we dissected key pre- and post-synaptic components with pharmacological strategies. We identified a significant and selective increase in post-synaptic nicotinic receptor signalling on prefrontal cortical neurons. To probe the additional impact of therapeutic intervention on the adapted circuit, we tested cholinergic and nicotinic-selective pro-cognitive treatments. Inhibition of acetylcholinesterase further enhanced endogenous cholinergic responses but greatly distorted their kinetics. Positive allosteric modulation of nicotinic receptors, by contrast, enhanced endogenous cholinergic responses and retained their rapid kinetics.
Conclusions: We demonstrate that functional nicotinic upregulation occurs within the prefrontal cortex in two AD models. Promisingly, this nicotinic signal can be further enhanced while preserving its rapid kinetic signature. Taken together, our work suggests that compensatory mechanisms are active within the prefrontal cortex that can be harnessed by nicotinic receptor positive allosteric modulation, highlighting a new direction for cognitive treatment in AD neuropathology.
期刊介绍:
Translational Neurodegeneration, an open-access, peer-reviewed journal, addresses all aspects of neurodegenerative diseases. It serves as a prominent platform for research, therapeutics, and education, fostering discussions and insights across basic, translational, and clinical research domains. Covering Parkinson's disease, Alzheimer's disease, and other neurodegenerative conditions, it welcomes contributions on epidemiology, pathogenesis, diagnosis, prevention, drug development, rehabilitation, and drug delivery. Scientists, clinicians, and physician-scientists are encouraged to share their work in this specialized journal tailored to their fields.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
