{"title":"新型GATA3突变p.a ala287asp引起的甲状旁腺功能减退、感音神经性耳聋和肾脏疾病(HDR)综合征","authors":"Luke Vroegindewey, John Kim, Dennis J Joseph","doi":"10.1530/EDM-24-0020","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>HDR is a rare autosomal dominant genetic disorder characterized by the triad of hypoparathyroidism, sensorineural deafness and renal anomalies caused by haploinsufficiency loss of function of the GATA-binding protein 3 (GATA3) gene. We present a case of a 56-year-old male diagnosed with hypoparathyroidism, sensorineural deafness, renal hypoplasia and epilepsy. Genetic testing revealed a novel GATA3 heterozygous mutation c.860C>A with a predicted amino acid substitution p.Ala287Asp. This hitherto unreported missense GATA mutation was characterized by a relatively late-onset and milder phenotype of the HDR triad.</p><p><strong>Learning points: </strong>GATA3 gene mutations located on chromosome 10p cause haploinsufficiency of the GATA3 protein affecting fetal development of the parathyroid glands, inner ear and renal anomalies, resulting in HDR syndrome with an autosomal dominant inheritance pattern.Also known as Barakat syndrome, it has been reported in less than 200 cases with an identified mutation, each having a varied phenotypic presentation without consistent genotypic correlation.We present a patient with HDR syndrome who tested positive for a novel mutation c.860C>A, resulting in a missense substitution of amino acids p.Ala287Asp in the GATA3 gene.Clinicians who identify this rare triad of hypoparathyroidism, sensorineural deafness and renal anomalies should further investigate with genetic testing for GATA3 mutations.</p>","PeriodicalId":37467,"journal":{"name":"Endocrinology, Diabetes and Metabolism Case Reports","volume":"2024 4","pages":""},"PeriodicalIF":0.7000,"publicationDate":"2024-12-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11737508/pdf/","citationCount":"0","resultStr":"{\"title\":\"Hypoparathyroidism, sensorineural deafness and renal disease (HDR) syndrome due to a novel GATA3 mutation p.Ala287Asp.\",\"authors\":\"Luke Vroegindewey, John Kim, Dennis J Joseph\",\"doi\":\"10.1530/EDM-24-0020\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Summary: </strong>HDR is a rare autosomal dominant genetic disorder characterized by the triad of hypoparathyroidism, sensorineural deafness and renal anomalies caused by haploinsufficiency loss of function of the GATA-binding protein 3 (GATA3) gene. We present a case of a 56-year-old male diagnosed with hypoparathyroidism, sensorineural deafness, renal hypoplasia and epilepsy. Genetic testing revealed a novel GATA3 heterozygous mutation c.860C>A with a predicted amino acid substitution p.Ala287Asp. This hitherto unreported missense GATA mutation was characterized by a relatively late-onset and milder phenotype of the HDR triad.</p><p><strong>Learning points: </strong>GATA3 gene mutations located on chromosome 10p cause haploinsufficiency of the GATA3 protein affecting fetal development of the parathyroid glands, inner ear and renal anomalies, resulting in HDR syndrome with an autosomal dominant inheritance pattern.Also known as Barakat syndrome, it has been reported in less than 200 cases with an identified mutation, each having a varied phenotypic presentation without consistent genotypic correlation.We present a patient with HDR syndrome who tested positive for a novel mutation c.860C>A, resulting in a missense substitution of amino acids p.Ala287Asp in the GATA3 gene.Clinicians who identify this rare triad of hypoparathyroidism, sensorineural deafness and renal anomalies should further investigate with genetic testing for GATA3 mutations.</p>\",\"PeriodicalId\":37467,\"journal\":{\"name\":\"Endocrinology, Diabetes and Metabolism Case Reports\",\"volume\":\"2024 4\",\"pages\":\"\"},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2024-12-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11737508/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Endocrinology, Diabetes and Metabolism Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1530/EDM-24-0020\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/1 0:00:00\",\"PubModel\":\"Print\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrinology, Diabetes and Metabolism Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1530/EDM-24-0020","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/1 0:00:00","PubModel":"Print","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Hypoparathyroidism, sensorineural deafness and renal disease (HDR) syndrome due to a novel GATA3 mutation p.Ala287Asp.
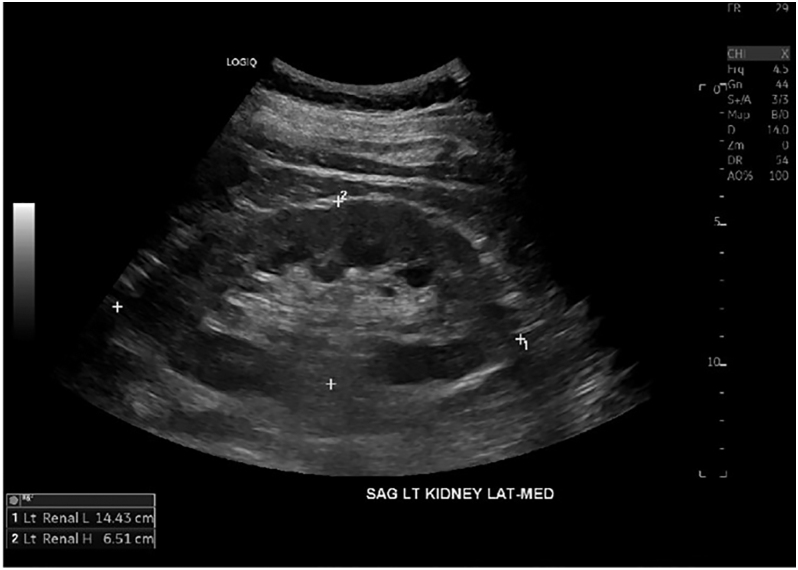
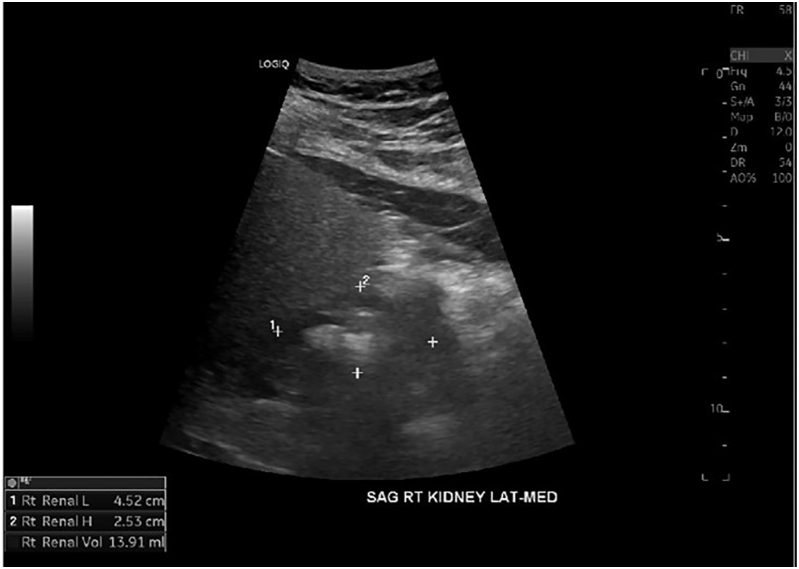
Summary: HDR is a rare autosomal dominant genetic disorder characterized by the triad of hypoparathyroidism, sensorineural deafness and renal anomalies caused by haploinsufficiency loss of function of the GATA-binding protein 3 (GATA3) gene. We present a case of a 56-year-old male diagnosed with hypoparathyroidism, sensorineural deafness, renal hypoplasia and epilepsy. Genetic testing revealed a novel GATA3 heterozygous mutation c.860C>A with a predicted amino acid substitution p.Ala287Asp. This hitherto unreported missense GATA mutation was characterized by a relatively late-onset and milder phenotype of the HDR triad.
Learning points: GATA3 gene mutations located on chromosome 10p cause haploinsufficiency of the GATA3 protein affecting fetal development of the parathyroid glands, inner ear and renal anomalies, resulting in HDR syndrome with an autosomal dominant inheritance pattern.Also known as Barakat syndrome, it has been reported in less than 200 cases with an identified mutation, each having a varied phenotypic presentation without consistent genotypic correlation.We present a patient with HDR syndrome who tested positive for a novel mutation c.860C>A, resulting in a missense substitution of amino acids p.Ala287Asp in the GATA3 gene.Clinicians who identify this rare triad of hypoparathyroidism, sensorineural deafness and renal anomalies should further investigate with genetic testing for GATA3 mutations.
期刊介绍:
Endocrinology, Diabetes & Metabolism Case Reports publishes case reports on common and rare conditions in all areas of clinical endocrinology, diabetes and metabolism. Articles should include clear learning points which readers can use to inform medical education or clinical practice. The types of cases of interest to Endocrinology, Diabetes & Metabolism Case Reports include: -Insight into disease pathogenesis or mechanism of therapy - Novel diagnostic procedure - Novel treatment - Unique/unexpected symptoms or presentations of a disease - New disease or syndrome: presentations/diagnosis/management - Unusual effects of medical treatment - Error in diagnosis/pitfalls and caveats





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
