Yaguo Gong, Yangbo Dai, Qibiao Wu, Li Guo, Xiaojun Yao, Qingxia Yang
{"title":"单细胞蛋白质组学数据整合的基准","authors":"Yaguo Gong, Yangbo Dai, Qibiao Wu, Li Guo, Xiaojun Yao, Qingxia Yang","doi":"10.1021/acs.analchem.4c04933","DOIUrl":null,"url":null,"abstract":"Single-cell proteomics (SCP) detected based on different technologies always involves batch-specific variations because of differences in sample processing and other potential biases. How to integrate SCP data effectively has become a great challenge. Integration of SCP data not only requires the conservation of true biological variances, but also realizes the removal of unwanted batch effects. In this study, benchmarking analysis of popular data integration methods was conducted to determine the most suitable method for SCP data. To comprehensively evaluate the performance of these integration methods, a novel evaluation system was proposed for integrating SCP data. This evaluation system consists of three objective measures from different perspectives: category (<i><b>a</b></i>), the efficacy of correcting batch effects; category (<i><b>b</b></i>), the power of conserving biological variances; and category (<i><b>c</b></i>), the ability to identify consistent markers. For this comprehensive evaluation, five benchmark data sets under different scenarios (containing substantial proteins, substantial cells, multiple batches, multiple cell types, and unbalanced data) were utilized for selecting the most suitable data integration method. As a result, three methods, ComBat, Scanorama, and Seurat version 3 CCA, were identified as the most recommended methods for integrating SCP data. Overall, this systematic evaluation might provide valuable guidance in choosing the appropriate method for data integration in the SCP.","PeriodicalId":27,"journal":{"name":"Analytical Chemistry","volume":"62 1","pages":""},"PeriodicalIF":6.7000,"publicationDate":"2025-01-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Benchmark of Data Integration in Single-Cell Proteomics\",\"authors\":\"Yaguo Gong, Yangbo Dai, Qibiao Wu, Li Guo, Xiaojun Yao, Qingxia Yang\",\"doi\":\"10.1021/acs.analchem.4c04933\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Single-cell proteomics (SCP) detected based on different technologies always involves batch-specific variations because of differences in sample processing and other potential biases. How to integrate SCP data effectively has become a great challenge. Integration of SCP data not only requires the conservation of true biological variances, but also realizes the removal of unwanted batch effects. In this study, benchmarking analysis of popular data integration methods was conducted to determine the most suitable method for SCP data. To comprehensively evaluate the performance of these integration methods, a novel evaluation system was proposed for integrating SCP data. This evaluation system consists of three objective measures from different perspectives: category (<i><b>a</b></i>), the efficacy of correcting batch effects; category (<i><b>b</b></i>), the power of conserving biological variances; and category (<i><b>c</b></i>), the ability to identify consistent markers. For this comprehensive evaluation, five benchmark data sets under different scenarios (containing substantial proteins, substantial cells, multiple batches, multiple cell types, and unbalanced data) were utilized for selecting the most suitable data integration method. As a result, three methods, ComBat, Scanorama, and Seurat version 3 CCA, were identified as the most recommended methods for integrating SCP data. Overall, this systematic evaluation might provide valuable guidance in choosing the appropriate method for data integration in the SCP.\",\"PeriodicalId\":27,\"journal\":{\"name\":\"Analytical Chemistry\",\"volume\":\"62 1\",\"pages\":\"\"},\"PeriodicalIF\":6.7000,\"publicationDate\":\"2025-01-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Analytical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.analchem.4c04933\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, ANALYTICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Analytical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.analchem.4c04933","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, ANALYTICAL","Score":null,"Total":0}
引用次数: 0
摘要
基于不同技术检测的单细胞蛋白质组学(SCP)由于样品处理的差异和其他潜在的偏差,总是存在批次特异性差异。如何有效地整合SCP数据已成为一个巨大的挑战。SCP数据的集成不仅要求保留真实的生物方差,而且要实现去除不需要的批效应。在本研究中,对常用的数据集成方法进行了基准分析,以确定最适合SCP数据的方法。为了综合评价这些集成方法的性能,提出了一种新的SCP数据集成评价体系。该评价体系由三个不同角度的客观指标组成:类别(a),纠正批次效果的有效性;(b)类,保存生物变异的能力;类别(c),识别一致标记的能力。为了进行综合评估,我们使用了5个不同场景下的基准数据集(包含大量蛋白质、大量细胞、多批次、多细胞类型和不平衡数据)来选择最合适的数据集成方法。因此,ComBat、Scanorama和Seurat version 3 CCA三种方法被认为是整合SCP数据的最推荐方法。总的来说,这种系统的评估可能为SCP中选择合适的数据集成方法提供有价值的指导。
Benchmark of Data Integration in Single-Cell Proteomics
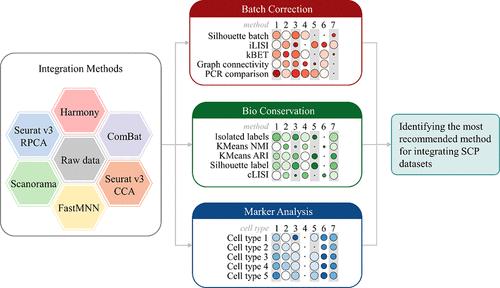
Single-cell proteomics (SCP) detected based on different technologies always involves batch-specific variations because of differences in sample processing and other potential biases. How to integrate SCP data effectively has become a great challenge. Integration of SCP data not only requires the conservation of true biological variances, but also realizes the removal of unwanted batch effects. In this study, benchmarking analysis of popular data integration methods was conducted to determine the most suitable method for SCP data. To comprehensively evaluate the performance of these integration methods, a novel evaluation system was proposed for integrating SCP data. This evaluation system consists of three objective measures from different perspectives: category (a), the efficacy of correcting batch effects; category (b), the power of conserving biological variances; and category (c), the ability to identify consistent markers. For this comprehensive evaluation, five benchmark data sets under different scenarios (containing substantial proteins, substantial cells, multiple batches, multiple cell types, and unbalanced data) were utilized for selecting the most suitable data integration method. As a result, three methods, ComBat, Scanorama, and Seurat version 3 CCA, were identified as the most recommended methods for integrating SCP data. Overall, this systematic evaluation might provide valuable guidance in choosing the appropriate method for data integration in the SCP.
期刊介绍:
Analytical Chemistry, a peer-reviewed research journal, focuses on disseminating new and original knowledge across all branches of analytical chemistry. Fundamental articles may explore general principles of chemical measurement science and need not directly address existing or potential analytical methodology. They can be entirely theoretical or report experimental results. Contributions may cover various phases of analytical operations, including sampling, bioanalysis, electrochemistry, mass spectrometry, microscale and nanoscale systems, environmental analysis, separations, spectroscopy, chemical reactions and selectivity, instrumentation, imaging, surface analysis, and data processing. Papers discussing known analytical methods should present a significant, original application of the method, a notable improvement, or results on an important analyte.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
