Yoonha Lee, Young Ah Lee, Jung Min Ko, Choong Ho Shin, Yun Jeong Lee
{"title":"儿童期先天性合并垂体激素缺乏症的临床和遗传特征:一项回顾性单中心队列研究","authors":"Yoonha Lee, Young Ah Lee, Jung Min Ko, Choong Ho Shin, Yun Jeong Lee","doi":"10.6065/apem.2448008.004","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>To investigate the clinical characteristics and genetic features of childhood-onset congenital combined pituitary hormone deficiency (cCPHD) in Korean patients.</p><p><strong>Methods: </strong>We retrospectively analyzed 444 patients diagnosed with childhood-onset CPHD at a tertiary center between 1994 and 2021. After excluding acquired case, 43 patients with cCPHD were enrolled. Anthropometric measurements, hormone evaluations, brain magnetic resonance imaging (MRI), extrapituitary phenotypes, and adult outcomes were analyzed. Genetic analyses were performed on 26 patients using a targeted gene panel or whole exome sequencing.</p><p><strong>Results: </strong>Mean age at diagnosis was 3.2 years, and 41.9% were diagnosed at less than 1 year old. Short stature was the most frequent (37.2%) initial presentation, and mean height z-score was -2.4. More than half (n=23, 53.5%) of patients had neonatal features suggestive of hypopituitarism; however, only 15 (65.2%) were diagnosed in infancy. Growth hormone deficiency (GHD) was prevalent in 42 (97.7%), and 33 (76.7%) had 3 or more hormone deficiencies. Extrapituitary phenotypes were identified in 31 (72.1%). Brain MRI abnormalities correlated with a higher number of hormone deficiencies (P for trend 0.049) and were present in 33 patients (80.5%). Adult GHD was diagnosed in all 17 investigated patients, and metabolic disturbances were noted in 10 (58.9%). Pathogenic variants in POU1F1, GLI2, HESX1, TBC1D32, and ROBO1 were found in 5 (19.2%).</p><p><strong>Conclusion: </strong>Considering the high proportion of neonatal presentations, identification of the early neonatal features of hypopituitarism to manage pituitary and extrapituitary phenotypes is critical. The genetic etiology of cCPHD warrants further exploration.</p>","PeriodicalId":44915,"journal":{"name":"Annals of Pediatric Endocrinology & Metabolism","volume":"29 6","pages":"379-386"},"PeriodicalIF":3.3000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11725638/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical and genetic features of childhood-onset congenital combined pituitary hormone deficiency: a retrospective, single-center cohort study.\",\"authors\":\"Yoonha Lee, Young Ah Lee, Jung Min Ko, Choong Ho Shin, Yun Jeong Lee\",\"doi\":\"10.6065/apem.2448008.004\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Purpose: </strong>To investigate the clinical characteristics and genetic features of childhood-onset congenital combined pituitary hormone deficiency (cCPHD) in Korean patients.</p><p><strong>Methods: </strong>We retrospectively analyzed 444 patients diagnosed with childhood-onset CPHD at a tertiary center between 1994 and 2021. After excluding acquired case, 43 patients with cCPHD were enrolled. Anthropometric measurements, hormone evaluations, brain magnetic resonance imaging (MRI), extrapituitary phenotypes, and adult outcomes were analyzed. Genetic analyses were performed on 26 patients using a targeted gene panel or whole exome sequencing.</p><p><strong>Results: </strong>Mean age at diagnosis was 3.2 years, and 41.9% were diagnosed at less than 1 year old. Short stature was the most frequent (37.2%) initial presentation, and mean height z-score was -2.4. More than half (n=23, 53.5%) of patients had neonatal features suggestive of hypopituitarism; however, only 15 (65.2%) were diagnosed in infancy. Growth hormone deficiency (GHD) was prevalent in 42 (97.7%), and 33 (76.7%) had 3 or more hormone deficiencies. Extrapituitary phenotypes were identified in 31 (72.1%). Brain MRI abnormalities correlated with a higher number of hormone deficiencies (P for trend 0.049) and were present in 33 patients (80.5%). Adult GHD was diagnosed in all 17 investigated patients, and metabolic disturbances were noted in 10 (58.9%). Pathogenic variants in POU1F1, GLI2, HESX1, TBC1D32, and ROBO1 were found in 5 (19.2%).</p><p><strong>Conclusion: </strong>Considering the high proportion of neonatal presentations, identification of the early neonatal features of hypopituitarism to manage pituitary and extrapituitary phenotypes is critical. The genetic etiology of cCPHD warrants further exploration.</p>\",\"PeriodicalId\":44915,\"journal\":{\"name\":\"Annals of Pediatric Endocrinology & Metabolism\",\"volume\":\"29 6\",\"pages\":\"379-386\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11725638/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Pediatric Endocrinology & Metabolism\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.6065/apem.2448008.004\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Pediatric Endocrinology & Metabolism","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.6065/apem.2448008.004","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/31 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Clinical and genetic features of childhood-onset congenital combined pituitary hormone deficiency: a retrospective, single-center cohort study.
Purpose: To investigate the clinical characteristics and genetic features of childhood-onset congenital combined pituitary hormone deficiency (cCPHD) in Korean patients.
Methods: We retrospectively analyzed 444 patients diagnosed with childhood-onset CPHD at a tertiary center between 1994 and 2021. After excluding acquired case, 43 patients with cCPHD were enrolled. Anthropometric measurements, hormone evaluations, brain magnetic resonance imaging (MRI), extrapituitary phenotypes, and adult outcomes were analyzed. Genetic analyses were performed on 26 patients using a targeted gene panel or whole exome sequencing.
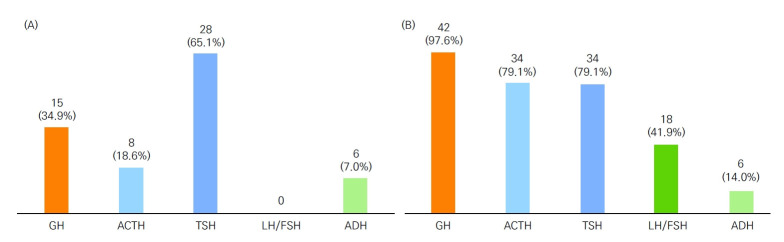
Results: Mean age at diagnosis was 3.2 years, and 41.9% were diagnosed at less than 1 year old. Short stature was the most frequent (37.2%) initial presentation, and mean height z-score was -2.4. More than half (n=23, 53.5%) of patients had neonatal features suggestive of hypopituitarism; however, only 15 (65.2%) were diagnosed in infancy. Growth hormone deficiency (GHD) was prevalent in 42 (97.7%), and 33 (76.7%) had 3 or more hormone deficiencies. Extrapituitary phenotypes were identified in 31 (72.1%). Brain MRI abnormalities correlated with a higher number of hormone deficiencies (P for trend 0.049) and were present in 33 patients (80.5%). Adult GHD was diagnosed in all 17 investigated patients, and metabolic disturbances were noted in 10 (58.9%). Pathogenic variants in POU1F1, GLI2, HESX1, TBC1D32, and ROBO1 were found in 5 (19.2%).
Conclusion: Considering the high proportion of neonatal presentations, identification of the early neonatal features of hypopituitarism to manage pituitary and extrapituitary phenotypes is critical. The genetic etiology of cCPHD warrants further exploration.
期刊介绍:
The Annals of Pediatric Endocrinology & Metabolism Journal is the official publication of the Korean Society of Pediatric Endocrinology. Its formal abbreviated title is “Ann Pediatr Endocrinol Metab”. It is a peer-reviewed open access journal of medicine published in English. The journal was launched in 1996 under the title of ‘Journal of Korean Society of Pediatric Endocrinology’ until 2011 (pISSN 1226-2242). Since 2012, the title is now changed to ‘Annals of Pediatric Endocrinology & Metabolism’. The Journal is published four times per year on the last day of March, June, September, and December. It is widely distributed for free to members of the Korean Society of Pediatric Endocrinology, medical schools, libraries, and academic institutions. The journal is indexed/tracked/covered by web sites of PubMed Central, PubMed, Emerging Sources Citation Index (ESCI), Scopus, EBSCO, EMBASE, KoreaMed, KoMCI, KCI, Science Central, DOI/CrossRef, Directory of Open Access Journals(DOAJ), and Google Scholar. The aims of Annals of Pediatric Endocrinology & Metabolism are to contribute to the advancements in the fields of pediatric endocrinology & metabolism through the scientific reviews and interchange of all of pediatric endocrinology and metabolism. It aims to reflect the latest clinical, translational, and basic research trends from worldwide valuable achievements. In addition, genome research, epidemiology, public education and clinical practice guidelines in each country are welcomed for publication. The Journal particularly focuses on research conducted with Asian-Pacific children whose genetic and environmental backgrounds are different from those of the Western. Area of specific interest include the following : Growth, puberty, glucose metabolism including diabetes mellitus, obesity, nutrition, disorders of sexual development, pituitary, thyroid, parathyroid, adrenal cortex, bone or other endocrine and metabolic disorders from infancy through adolescence.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
