Kit Joll, Philipp Schienbein, Kevin M. Rosso, Jochen Blumberger
{"title":"反应神经网络电位分子动力学揭示铁(II)在赤铁矿(001)上的化学吸附机理","authors":"Kit Joll, Philipp Schienbein, Kevin M. Rosso, Jochen Blumberger","doi":"10.1021/acs.jpclett.4c03252","DOIUrl":null,"url":null,"abstract":"Atomic-scale understanding of important geochemical processes including sorption, dissolution, nucleation, and crystal growth is difficult to obtain from experimental measurements alone and would benefit from strong continuous progress in molecular simulation. To this end, we present a reactive neural network potential-based molecular dynamics approach to simulate the interaction of aqueous ions on mineral surfaces in contact with liquid water, taking Fe(II) on hematite(001) as a model system. We show that a single neural network potential predicts rate constants for water exchange for aqueous Fe(II) and for the exergonic chemisorption of aqueous Fe(II) on hematite(001) in good agreement with experimental observations. The neural network potential developed herein allows one to converge free energy profiles and transmission coefficients at density functional theory-level accuracy outperforming state-of-the-art classical force field potentials. This suggests that machine learning potential molecular dynamics should become the method of choice for atomistic studies of geochemical processes.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"41 1","pages":""},"PeriodicalIF":4.5000,"publicationDate":"2025-01-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mechanism of Fe(II) Chemisorption on Hematite(001) Revealed by Reactive Neural Network Potential Molecular Dynamics\",\"authors\":\"Kit Joll, Philipp Schienbein, Kevin M. Rosso, Jochen Blumberger\",\"doi\":\"10.1021/acs.jpclett.4c03252\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Atomic-scale understanding of important geochemical processes including sorption, dissolution, nucleation, and crystal growth is difficult to obtain from experimental measurements alone and would benefit from strong continuous progress in molecular simulation. To this end, we present a reactive neural network potential-based molecular dynamics approach to simulate the interaction of aqueous ions on mineral surfaces in contact with liquid water, taking Fe(II) on hematite(001) as a model system. We show that a single neural network potential predicts rate constants for water exchange for aqueous Fe(II) and for the exergonic chemisorption of aqueous Fe(II) on hematite(001) in good agreement with experimental observations. The neural network potential developed herein allows one to converge free energy profiles and transmission coefficients at density functional theory-level accuracy outperforming state-of-the-art classical force field potentials. This suggests that machine learning potential molecular dynamics should become the method of choice for atomistic studies of geochemical processes.\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"41 1\",\"pages\":\"\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2025-01-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpclett.4c03252\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c03252","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
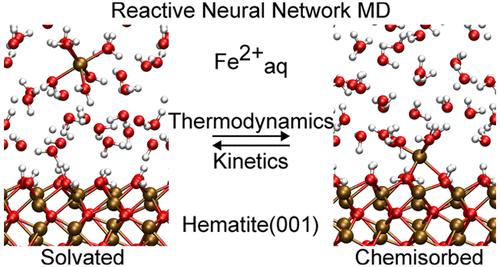
Mechanism of Fe(II) Chemisorption on Hematite(001) Revealed by Reactive Neural Network Potential Molecular Dynamics
Atomic-scale understanding of important geochemical processes including sorption, dissolution, nucleation, and crystal growth is difficult to obtain from experimental measurements alone and would benefit from strong continuous progress in molecular simulation. To this end, we present a reactive neural network potential-based molecular dynamics approach to simulate the interaction of aqueous ions on mineral surfaces in contact with liquid water, taking Fe(II) on hematite(001) as a model system. We show that a single neural network potential predicts rate constants for water exchange for aqueous Fe(II) and for the exergonic chemisorption of aqueous Fe(II) on hematite(001) in good agreement with experimental observations. The neural network potential developed herein allows one to converge free energy profiles and transmission coefficients at density functional theory-level accuracy outperforming state-of-the-art classical force field potentials. This suggests that machine learning potential molecular dynamics should become the method of choice for atomistic studies of geochemical processes.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
