{"title":"了解单原子掺杂Cu/Cu2O催化剂对乙醇的CO2还原选择性:来自Bader电荷描述符的见解","authors":"Yunchen Qian, Jinshan Liang, Lijuan Xie, Ke Zheng, Weixin Lin, Lizhi Jiang","doi":"10.1021/acs.jpclett.4c03269","DOIUrl":null,"url":null,"abstract":"In the CO<sub>2</sub> reduction reactions (CO<sub>2</sub>RR), the product selectivity is strongly dependent on the binding energy differences of the key intermediates. Herein, we systematically evaluated the CO<sub>2</sub>RR reaction pathways on single transition metal atom doped catalysts TM<sub>1</sub>Cu/Cu<sub>2</sub>O by density functional theory (DFT) methods and found that *CO is more likely to undergo C–O bond cleavage rather than be hydrogenated on TM<sub>1</sub>Cu/Cu<sub>2</sub>O (TM = Sc, Ti, V, Cr, Mn, Fe, Co), which facilitates C<sub>2+</sub> production with a low-energy pathway of OC–C coupling, while it prefers to be hydrogenated to form CHO on TM<sub>1</sub>Cu/Cu<sub>2</sub>O (TM = Ni, Cu). The defects of Cu in TM<sub>1</sub>Cu/Cu<sub>2</sub>O were confirmed to enhance the production of ethanol. Furthermore, we established a scaling relationship between binding free energies of the key intermediates with the Bader charges of the active sites TM on TM<sub>1</sub>Cu/Cu<sub>2</sub>O and defective TM<sub>1</sub>Cu/Cu<sub>2</sub>O surfaces. This relationship facilitates a rational and efficient design of Cu/Cu<sub>2</sub>O-based catalysts.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"12 1","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2025-01-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Understanding the CO2 Reduction Selectivity toward Ethanol on Single Atom Doped Cu/Cu2O Catalysts: Insights from Bader Charge as a Descriptor\",\"authors\":\"Yunchen Qian, Jinshan Liang, Lijuan Xie, Ke Zheng, Weixin Lin, Lizhi Jiang\",\"doi\":\"10.1021/acs.jpclett.4c03269\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"In the CO<sub>2</sub> reduction reactions (CO<sub>2</sub>RR), the product selectivity is strongly dependent on the binding energy differences of the key intermediates. Herein, we systematically evaluated the CO<sub>2</sub>RR reaction pathways on single transition metal atom doped catalysts TM<sub>1</sub>Cu/Cu<sub>2</sub>O by density functional theory (DFT) methods and found that *CO is more likely to undergo C–O bond cleavage rather than be hydrogenated on TM<sub>1</sub>Cu/Cu<sub>2</sub>O (TM = Sc, Ti, V, Cr, Mn, Fe, Co), which facilitates C<sub>2+</sub> production with a low-energy pathway of OC–C coupling, while it prefers to be hydrogenated to form CHO on TM<sub>1</sub>Cu/Cu<sub>2</sub>O (TM = Ni, Cu). The defects of Cu in TM<sub>1</sub>Cu/Cu<sub>2</sub>O were confirmed to enhance the production of ethanol. Furthermore, we established a scaling relationship between binding free energies of the key intermediates with the Bader charges of the active sites TM on TM<sub>1</sub>Cu/Cu<sub>2</sub>O and defective TM<sub>1</sub>Cu/Cu<sub>2</sub>O surfaces. This relationship facilitates a rational and efficient design of Cu/Cu<sub>2</sub>O-based catalysts.\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"12 1\",\"pages\":\"\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-01-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpclett.4c03269\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c03269","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Understanding the CO2 Reduction Selectivity toward Ethanol on Single Atom Doped Cu/Cu2O Catalysts: Insights from Bader Charge as a Descriptor
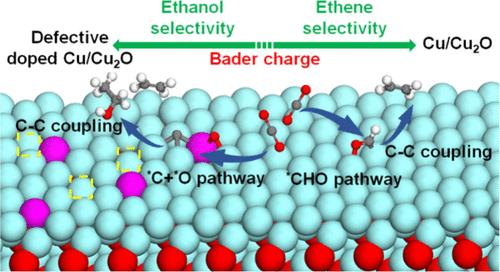
In the CO2 reduction reactions (CO2RR), the product selectivity is strongly dependent on the binding energy differences of the key intermediates. Herein, we systematically evaluated the CO2RR reaction pathways on single transition metal atom doped catalysts TM1Cu/Cu2O by density functional theory (DFT) methods and found that *CO is more likely to undergo C–O bond cleavage rather than be hydrogenated on TM1Cu/Cu2O (TM = Sc, Ti, V, Cr, Mn, Fe, Co), which facilitates C2+ production with a low-energy pathway of OC–C coupling, while it prefers to be hydrogenated to form CHO on TM1Cu/Cu2O (TM = Ni, Cu). The defects of Cu in TM1Cu/Cu2O were confirmed to enhance the production of ethanol. Furthermore, we established a scaling relationship between binding free energies of the key intermediates with the Bader charges of the active sites TM on TM1Cu/Cu2O and defective TM1Cu/Cu2O surfaces. This relationship facilitates a rational and efficient design of Cu/Cu2O-based catalysts.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
