Lukas Hasecke, Maximilian Breitenbach, Martí Gimferrer, Rainer Oswald, Ricardo A Mata
{"title":"用密度拟合多分量密度泛函理论求解非调和效应。","authors":"Lukas Hasecke, Maximilian Breitenbach, Martí Gimferrer, Rainer Oswald, Ricardo A Mata","doi":"10.1021/acs.jpca.5c00382","DOIUrl":null,"url":null,"abstract":"<p><p>In this contribution we present the first local density-fitted multicomponent density functional theory implementation and assess its use for the calculation of anharmonic zero-point energies. Four challenging cases of molecular aggregates are reviewed: deprotonated formic acid trimer, diphenyl ether-<i>tert</i>-butyl alcohol conformers, anisole/methanol and anisole/2-naphtol dimers. These are all cases where a mismatch between the low-temperature computationally predicted minimum and the experimentally determined structure was observed. Through the use of nuclear-electronic orbital energies in the thermodynamic correction, the correct energetic ordering is recovered. For the smallest system, we compare our results to vibrational perturbation theory anharmonically corrected zero-point energy, with perfect agreement for the lower-lying conformers. The performance of the newly developed code and the density fitting errors are also analyzed. Overall, the new implementation shows a very good scaling with system size and the density fitting approximations exhibit a negligible impact.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":" ","pages":"3560-3566"},"PeriodicalIF":3.0000,"publicationDate":"2025-04-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12010317/pdf/","citationCount":"0","resultStr":"{\"title\":\"Addressing Anharmonic Effects with Density-Fitted Multicomponent Density Functional Theory.\",\"authors\":\"Lukas Hasecke, Maximilian Breitenbach, Martí Gimferrer, Rainer Oswald, Ricardo A Mata\",\"doi\":\"10.1021/acs.jpca.5c00382\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>In this contribution we present the first local density-fitted multicomponent density functional theory implementation and assess its use for the calculation of anharmonic zero-point energies. Four challenging cases of molecular aggregates are reviewed: deprotonated formic acid trimer, diphenyl ether-<i>tert</i>-butyl alcohol conformers, anisole/methanol and anisole/2-naphtol dimers. These are all cases where a mismatch between the low-temperature computationally predicted minimum and the experimentally determined structure was observed. Through the use of nuclear-electronic orbital energies in the thermodynamic correction, the correct energetic ordering is recovered. For the smallest system, we compare our results to vibrational perturbation theory anharmonically corrected zero-point energy, with perfect agreement for the lower-lying conformers. The performance of the newly developed code and the density fitting errors are also analyzed. Overall, the new implementation shows a very good scaling with system size and the density fitting approximations exhibit a negligible impact.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\" \",\"pages\":\"3560-3566\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2025-04-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12010317/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpca.5c00382\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/4/7 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.5c00382","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/7 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Addressing Anharmonic Effects with Density-Fitted Multicomponent Density Functional Theory.
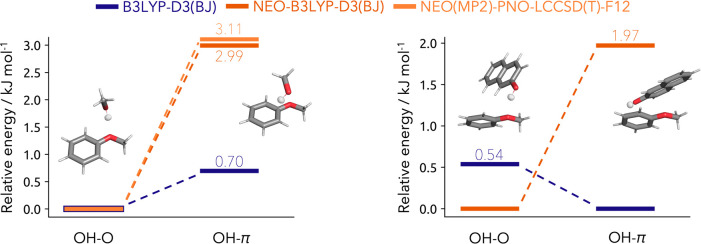
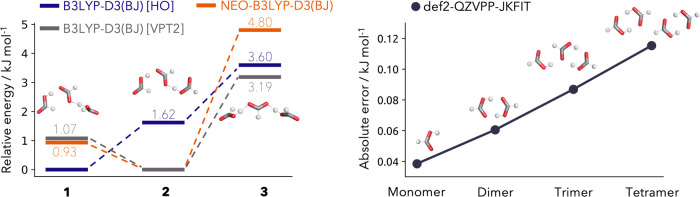
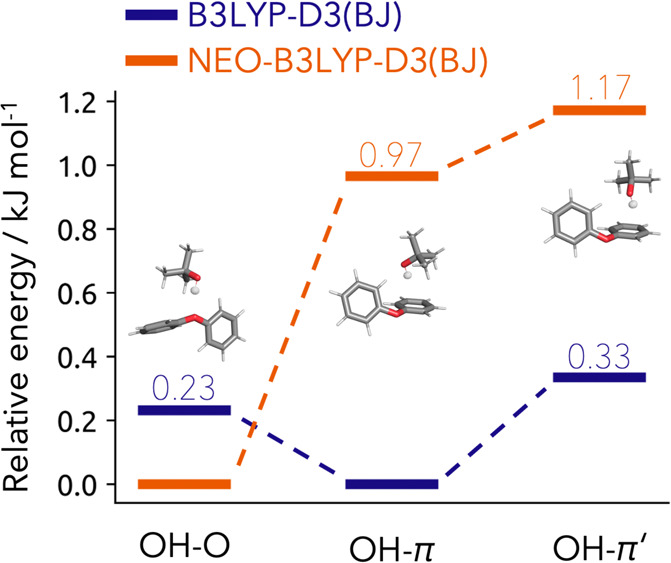
In this contribution we present the first local density-fitted multicomponent density functional theory implementation and assess its use for the calculation of anharmonic zero-point energies. Four challenging cases of molecular aggregates are reviewed: deprotonated formic acid trimer, diphenyl ether-tert-butyl alcohol conformers, anisole/methanol and anisole/2-naphtol dimers. These are all cases where a mismatch between the low-temperature computationally predicted minimum and the experimentally determined structure was observed. Through the use of nuclear-electronic orbital energies in the thermodynamic correction, the correct energetic ordering is recovered. For the smallest system, we compare our results to vibrational perturbation theory anharmonically corrected zero-point energy, with perfect agreement for the lower-lying conformers. The performance of the newly developed code and the density fitting errors are also analyzed. Overall, the new implementation shows a very good scaling with system size and the density fitting approximations exhibit a negligible impact.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
