{"title":"ReSeq模拟真实的Illumina高通量测序数据。","authors":"Stephan Schmeing, Mark D Robinson","doi":"10.1186/s13059-021-02265-7","DOIUrl":null,"url":null,"abstract":"<p><p>In high-throughput sequencing data, performance comparisons between computational tools are essential for making informed decisions at each step of a project. Simulations are a critical part of method comparisons, but for standard Illumina sequencing of genomic DNA, they are often oversimplified, which leads to optimistic results for most tools. ReSeq improves the authenticity of synthetic data by extracting and reproducing key components from real data. Major advancements are the inclusion of systematic errors, a fragment-based coverage model and sampling-matrix estimates based on two-dimensional margins. These improvements lead to more faithful performance evaluations. ReSeq is available at https://github.com/schmeing/ReSeq .</p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"22 1","pages":"67"},"PeriodicalIF":9.2000,"publicationDate":"2021-02-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13059-021-02265-7","citationCount":"5","resultStr":"{\"title\":\"ReSeq simulates realistic Illumina high-throughput sequencing data.\",\"authors\":\"Stephan Schmeing, Mark D Robinson\",\"doi\":\"10.1186/s13059-021-02265-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>In high-throughput sequencing data, performance comparisons between computational tools are essential for making informed decisions at each step of a project. Simulations are a critical part of method comparisons, but for standard Illumina sequencing of genomic DNA, they are often oversimplified, which leads to optimistic results for most tools. ReSeq improves the authenticity of synthetic data by extracting and reproducing key components from real data. Major advancements are the inclusion of systematic errors, a fragment-based coverage model and sampling-matrix estimates based on two-dimensional margins. These improvements lead to more faithful performance evaluations. ReSeq is available at https://github.com/schmeing/ReSeq .</p>\",\"PeriodicalId\":48922,\"journal\":{\"name\":\"Genome Biology\",\"volume\":\"22 1\",\"pages\":\"67\"},\"PeriodicalIF\":9.2000,\"publicationDate\":\"2021-02-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13059-021-02265-7\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13059-021-02265-7\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-021-02265-7","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
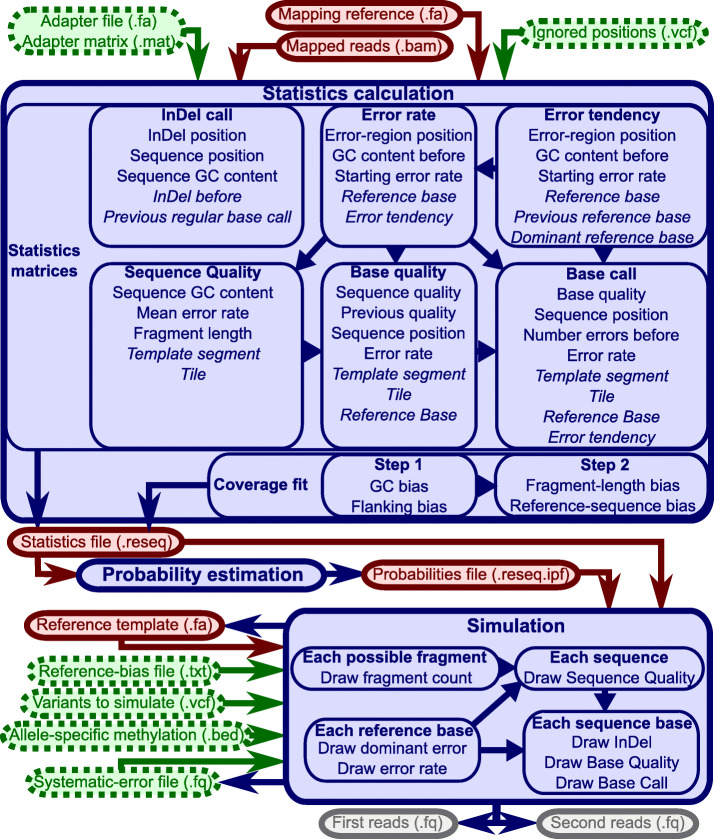
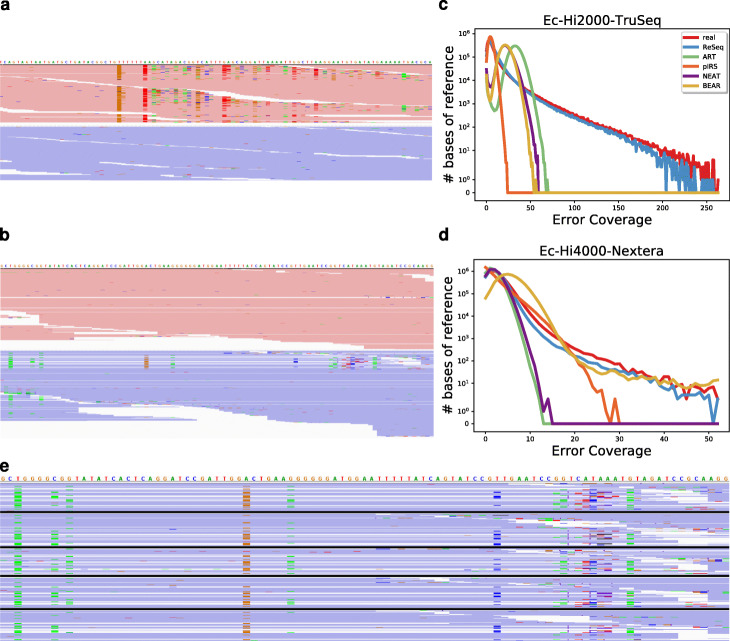
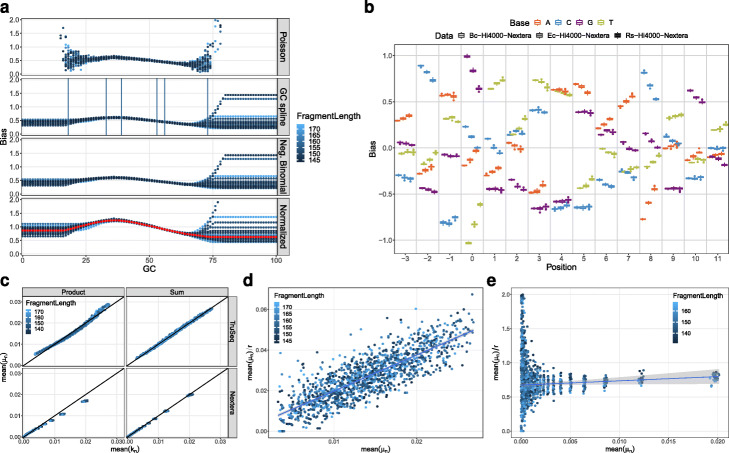
In high-throughput sequencing data, performance comparisons between computational tools are essential for making informed decisions at each step of a project. Simulations are a critical part of method comparisons, but for standard Illumina sequencing of genomic DNA, they are often oversimplified, which leads to optimistic results for most tools. ReSeq improves the authenticity of synthetic data by extracting and reproducing key components from real data. Major advancements are the inclusion of systematic errors, a fragment-based coverage model and sampling-matrix estimates based on two-dimensional margins. These improvements lead to more faithful performance evaluations. ReSeq is available at https://github.com/schmeing/ReSeq .
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
