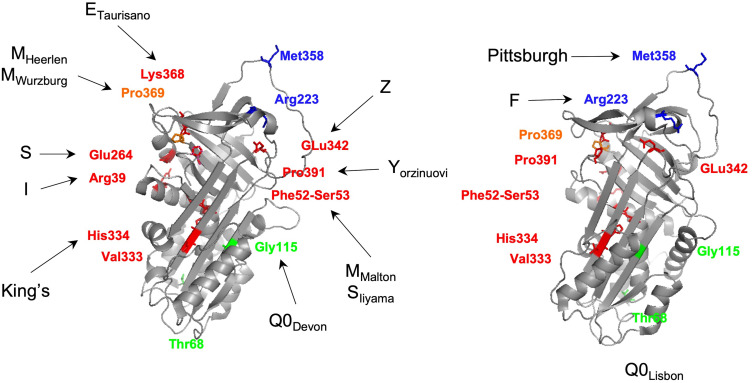
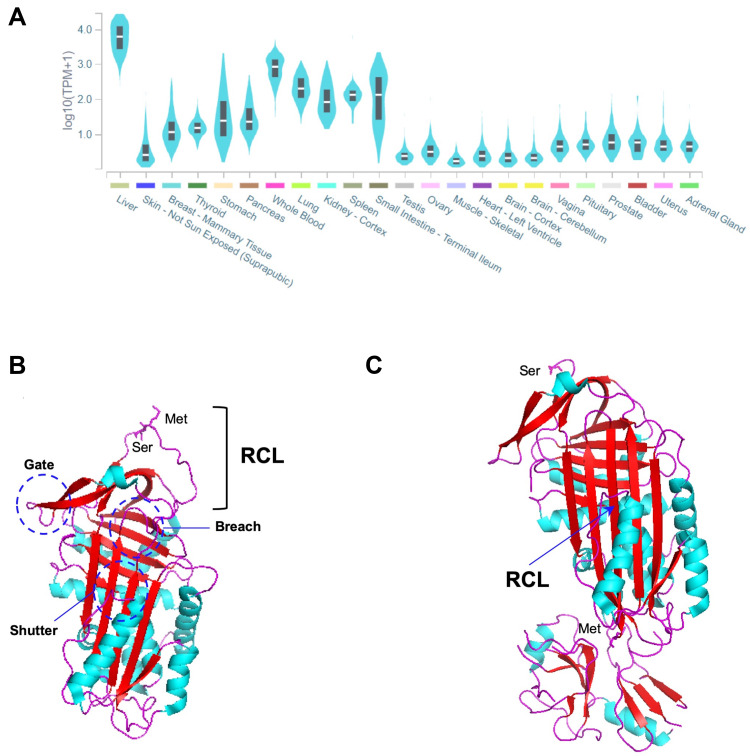
{"title":"α -1抗胰蛋白酶缺乏症的已知突变:SERPINA1变异谱的最新概述。","authors":"Susana Seixas, Patricia Isabel Marques","doi":"10.2147/TACG.S257511","DOIUrl":null,"url":null,"abstract":"<p><p>Alpha-1-Antitrypsin deficiency (AATD), caused by <i>SERPINA1</i> mutations, is one of the most prevalent Mendelian disorders among individuals of European descend. However, this condition, which is characterized by reduced serum levels of alpha-1-antitrypsin (AAT) and associated with increased risks of pulmonary emphysema and liver disease in both children and adults, remains frequently underdiagnosed. AATD clinical manifestations are often correlated with two pathogenic variants, the Z allele (p.Glu342Lys) and the S allele (p.Glu264Val), which can be combined in severe ZZ or moderate SZ risk genotypes. Yet, screenings of AATD cases and large sequencing efforts carried out in both control and disease populations are disclosing outstanding numbers of rare <i>SERPINA1</i> variants (>500), including many pathogenic and other likely deleterious mutations. Generally speaking, pathogenic variants can be subdivided into either loss- or gain-of-function according to their pathophysiological effects. In AATD, the loss-of-function is correlated with an uncontrolled activity of elastase by its natural inhibitor, the AAT. This phenomenon can result from the absence of circulating AAT (null alleles), poor AAT secretion from hepatocytes (deficiency alleles) or even from a modified inhibitory activity (dysfunctional alleles). On the other hand, the gain-of-function is connected with the formation of AAT polymers and their switching on of cellular stress and inflammatory responses (deficiency alleles). Less frequently, the gain-of-function is related to a modified protease affinity (dysfunctional alleles). Here, we revisit <i>SERPINA1</i> mutation spectrum, its origins and population history with a greater emphasis on variants fitting the aforementioned processes of AATD pathogenesis. Those were selected based on their clinical significance and wider geographic distribution. Moreover, we also provide some directions for future studies of AATD clinically heterogeneity and comprehensive diagnosis.</p>","PeriodicalId":506374,"journal":{"name":"The Application of Clinical Genetics","volume":"14 ","pages":"173-194"},"PeriodicalIF":0.0000,"publicationDate":"2021-03-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6e/4d/tacg-14-173.PMC7997584.pdf","citationCount":"36","resultStr":"{\"title\":\"Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of <i>SERPINA1</i> Variation Spectrum.\",\"authors\":\"Susana Seixas, Patricia Isabel Marques\",\"doi\":\"10.2147/TACG.S257511\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Alpha-1-Antitrypsin deficiency (AATD), caused by <i>SERPINA1</i> mutations, is one of the most prevalent Mendelian disorders among individuals of European descend. However, this condition, which is characterized by reduced serum levels of alpha-1-antitrypsin (AAT) and associated with increased risks of pulmonary emphysema and liver disease in both children and adults, remains frequently underdiagnosed. AATD clinical manifestations are often correlated with two pathogenic variants, the Z allele (p.Glu342Lys) and the S allele (p.Glu264Val), which can be combined in severe ZZ or moderate SZ risk genotypes. Yet, screenings of AATD cases and large sequencing efforts carried out in both control and disease populations are disclosing outstanding numbers of rare <i>SERPINA1</i> variants (>500), including many pathogenic and other likely deleterious mutations. Generally speaking, pathogenic variants can be subdivided into either loss- or gain-of-function according to their pathophysiological effects. In AATD, the loss-of-function is correlated with an uncontrolled activity of elastase by its natural inhibitor, the AAT. This phenomenon can result from the absence of circulating AAT (null alleles), poor AAT secretion from hepatocytes (deficiency alleles) or even from a modified inhibitory activity (dysfunctional alleles). On the other hand, the gain-of-function is connected with the formation of AAT polymers and their switching on of cellular stress and inflammatory responses (deficiency alleles). Less frequently, the gain-of-function is related to a modified protease affinity (dysfunctional alleles). Here, we revisit <i>SERPINA1</i> mutation spectrum, its origins and population history with a greater emphasis on variants fitting the aforementioned processes of AATD pathogenesis. Those were selected based on their clinical significance and wider geographic distribution. Moreover, we also provide some directions for future studies of AATD clinically heterogeneity and comprehensive diagnosis.</p>\",\"PeriodicalId\":506374,\"journal\":{\"name\":\"The Application of Clinical Genetics\",\"volume\":\"14 \",\"pages\":\"173-194\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-03-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6e/4d/tacg-14-173.PMC7997584.pdf\",\"citationCount\":\"36\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Application of Clinical Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/TACG.S257511\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Application of Clinical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S257511","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum.
Alpha-1-Antitrypsin deficiency (AATD), caused by SERPINA1 mutations, is one of the most prevalent Mendelian disorders among individuals of European descend. However, this condition, which is characterized by reduced serum levels of alpha-1-antitrypsin (AAT) and associated with increased risks of pulmonary emphysema and liver disease in both children and adults, remains frequently underdiagnosed. AATD clinical manifestations are often correlated with two pathogenic variants, the Z allele (p.Glu342Lys) and the S allele (p.Glu264Val), which can be combined in severe ZZ or moderate SZ risk genotypes. Yet, screenings of AATD cases and large sequencing efforts carried out in both control and disease populations are disclosing outstanding numbers of rare SERPINA1 variants (>500), including many pathogenic and other likely deleterious mutations. Generally speaking, pathogenic variants can be subdivided into either loss- or gain-of-function according to their pathophysiological effects. In AATD, the loss-of-function is correlated with an uncontrolled activity of elastase by its natural inhibitor, the AAT. This phenomenon can result from the absence of circulating AAT (null alleles), poor AAT secretion from hepatocytes (deficiency alleles) or even from a modified inhibitory activity (dysfunctional alleles). On the other hand, the gain-of-function is connected with the formation of AAT polymers and their switching on of cellular stress and inflammatory responses (deficiency alleles). Less frequently, the gain-of-function is related to a modified protease affinity (dysfunctional alleles). Here, we revisit SERPINA1 mutation spectrum, its origins and population history with a greater emphasis on variants fitting the aforementioned processes of AATD pathogenesis. Those were selected based on their clinical significance and wider geographic distribution. Moreover, we also provide some directions for future studies of AATD clinically heterogeneity and comprehensive diagnosis.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
