{"title":"利用共振识别模型设计生物活性肽。","authors":"Irena Cosic, Elena Pirogova","doi":"10.1186/1753-4631-1-7","DOIUrl":null,"url":null,"abstract":"<p><p> With a large number of DNA and protein sequences already known, the crucial question is to find out how the biological function of these macromolecules is \"written\" in the sequence of nucleotides or amino acids. Biological processes in any living organism are based on selective interactions between particular bio-molecules, mostly proteins. The rules governing the coding of a protein's biological function, i.e. its ability to selectively interact with other molecules, are still not elucidated. In addition, with the rapid accumulation of databases of protein primary structures, there is an urgent need for theoretical approaches that are capable of analysing protein structure-function relationships. The Resonant Recognition Model (RRM) 12 is one attempt to identify the selectivity of protein interactions within the amino acid sequence. The RRM 12 is a physico-mathematical approach that interprets protein sequence linear information using digital signal processing methods. In the RRM the protein primary structure is represented as a numerical series by assigning to each amino acid in the sequence a physical parameter value relevant to the protein's biological activity. The RRM concept is based on the finding that there is a significant correlation between spectra of the numerical presentation of amino acids and their biological activity. Once the characteristic frequency for a particular protein function/interaction is identified, it is possible then to utilize the RRM approach to predict the amino acids in the protein sequence, which predominantly contribute to this frequency and thus, to the observed function, as well as to design de novo peptides having the desired periodicities. As was shown in our previous studies of fibroblast growth factor (FGF) peptidic antagonists 23 and human immunodeficiency virus (HIV) envelope agonists 24, such de novo designed peptides express desired biological function. This study utilises the RRM computational approach to the analysis of oncogene and proto-oncogene proteins. The results obtained have shown that the RRM is capable of identifying the differences between the oncogenic and proto-oncogenic proteins with the possibility of identifying the \"cancer-causing\" features within their protein primary structure. In addition, the rational design of bioactive peptide analogues displaying oncogenic or proto-oncogenic-like activity is presented here.</p>","PeriodicalId":87480,"journal":{"name":"Nonlinear biomedical physics","volume":"1 1","pages":"7"},"PeriodicalIF":0.0000,"publicationDate":"2007-07-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1753-4631-1-7","citationCount":"51","resultStr":"{\"title\":\"Bioactive peptide design using the Resonant Recognition Model.\",\"authors\":\"Irena Cosic, Elena Pirogova\",\"doi\":\"10.1186/1753-4631-1-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p> With a large number of DNA and protein sequences already known, the crucial question is to find out how the biological function of these macromolecules is \\\"written\\\" in the sequence of nucleotides or amino acids. Biological processes in any living organism are based on selective interactions between particular bio-molecules, mostly proteins. The rules governing the coding of a protein's biological function, i.e. its ability to selectively interact with other molecules, are still not elucidated. In addition, with the rapid accumulation of databases of protein primary structures, there is an urgent need for theoretical approaches that are capable of analysing protein structure-function relationships. The Resonant Recognition Model (RRM) 12 is one attempt to identify the selectivity of protein interactions within the amino acid sequence. The RRM 12 is a physico-mathematical approach that interprets protein sequence linear information using digital signal processing methods. In the RRM the protein primary structure is represented as a numerical series by assigning to each amino acid in the sequence a physical parameter value relevant to the protein's biological activity. The RRM concept is based on the finding that there is a significant correlation between spectra of the numerical presentation of amino acids and their biological activity. Once the characteristic frequency for a particular protein function/interaction is identified, it is possible then to utilize the RRM approach to predict the amino acids in the protein sequence, which predominantly contribute to this frequency and thus, to the observed function, as well as to design de novo peptides having the desired periodicities. As was shown in our previous studies of fibroblast growth factor (FGF) peptidic antagonists 23 and human immunodeficiency virus (HIV) envelope agonists 24, such de novo designed peptides express desired biological function. This study utilises the RRM computational approach to the analysis of oncogene and proto-oncogene proteins. The results obtained have shown that the RRM is capable of identifying the differences between the oncogenic and proto-oncogenic proteins with the possibility of identifying the \\\"cancer-causing\\\" features within their protein primary structure. In addition, the rational design of bioactive peptide analogues displaying oncogenic or proto-oncogenic-like activity is presented here.</p>\",\"PeriodicalId\":87480,\"journal\":{\"name\":\"Nonlinear biomedical physics\",\"volume\":\"1 1\",\"pages\":\"7\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2007-07-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1753-4631-1-7\",\"citationCount\":\"51\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nonlinear biomedical physics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1753-4631-1-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nonlinear biomedical physics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1753-4631-1-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Bioactive peptide design using the Resonant Recognition Model.
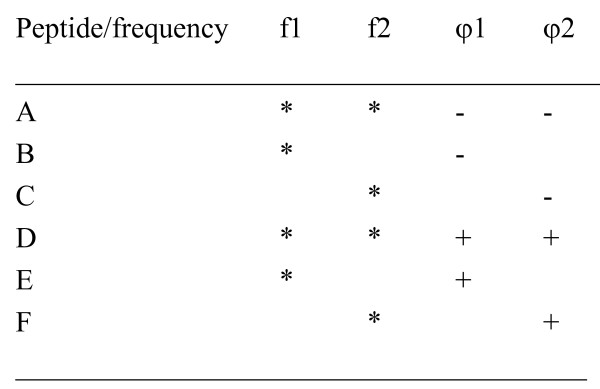
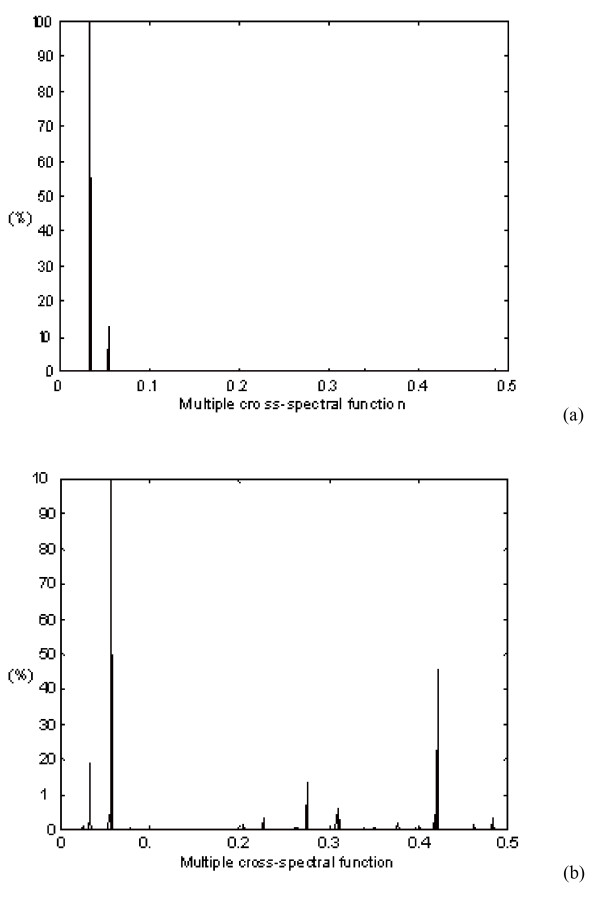
With a large number of DNA and protein sequences already known, the crucial question is to find out how the biological function of these macromolecules is "written" in the sequence of nucleotides or amino acids. Biological processes in any living organism are based on selective interactions between particular bio-molecules, mostly proteins. The rules governing the coding of a protein's biological function, i.e. its ability to selectively interact with other molecules, are still not elucidated. In addition, with the rapid accumulation of databases of protein primary structures, there is an urgent need for theoretical approaches that are capable of analysing protein structure-function relationships. The Resonant Recognition Model (RRM) 12 is one attempt to identify the selectivity of protein interactions within the amino acid sequence. The RRM 12 is a physico-mathematical approach that interprets protein sequence linear information using digital signal processing methods. In the RRM the protein primary structure is represented as a numerical series by assigning to each amino acid in the sequence a physical parameter value relevant to the protein's biological activity. The RRM concept is based on the finding that there is a significant correlation between spectra of the numerical presentation of amino acids and their biological activity. Once the characteristic frequency for a particular protein function/interaction is identified, it is possible then to utilize the RRM approach to predict the amino acids in the protein sequence, which predominantly contribute to this frequency and thus, to the observed function, as well as to design de novo peptides having the desired periodicities. As was shown in our previous studies of fibroblast growth factor (FGF) peptidic antagonists 23 and human immunodeficiency virus (HIV) envelope agonists 24, such de novo designed peptides express desired biological function. This study utilises the RRM computational approach to the analysis of oncogene and proto-oncogene proteins. The results obtained have shown that the RRM is capable of identifying the differences between the oncogenic and proto-oncogenic proteins with the possibility of identifying the "cancer-causing" features within their protein primary structure. In addition, the rational design of bioactive peptide analogues displaying oncogenic or proto-oncogenic-like activity is presented here.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
