Chris M Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B Maples, John Nemunaitis
{"title":"遗传性包涵体肌病(HIBM2)。","authors":"Chris M Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B Maples, John Nemunaitis","doi":"10.4137/grsb.s2594","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary inclusion body myopathy type 2 (HIBM2) is a myopathy characterized by progressive muscle weakness with early adult onset. The disease is the result of a recessive mutation in the Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase gene (GNE), which results in reduced enzyme function and sialic acid levels. A majority of individuals with HIBM2 are from Iranian-Jewish or Japanese decent, but isolated cases have been identified world wide. This article reviews the diagnostic criteria for HIBM2. Current research with a highlight on the biology of the disease and the role of GNE in the sialic acid pathway are assessed. Finally, therapeutic investigations and animal models are discussed with a focus on future studies to better understand the pathology of Hereditary Inclusion Body Myopathy and move therapeutic agents towards clinical trials.</p>","PeriodicalId":73138,"journal":{"name":"Gene regulation and systems biology","volume":"3 ","pages":"181-90"},"PeriodicalIF":0.0000,"publicationDate":"2009-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4137/grsb.s2594","citationCount":"13","resultStr":"{\"title\":\"Hereditary Inclusion Body Myopathy (HIBM2).\",\"authors\":\"Chris M Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B Maples, John Nemunaitis\",\"doi\":\"10.4137/grsb.s2594\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hereditary inclusion body myopathy type 2 (HIBM2) is a myopathy characterized by progressive muscle weakness with early adult onset. The disease is the result of a recessive mutation in the Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase gene (GNE), which results in reduced enzyme function and sialic acid levels. A majority of individuals with HIBM2 are from Iranian-Jewish or Japanese decent, but isolated cases have been identified world wide. This article reviews the diagnostic criteria for HIBM2. Current research with a highlight on the biology of the disease and the role of GNE in the sialic acid pathway are assessed. Finally, therapeutic investigations and animal models are discussed with a focus on future studies to better understand the pathology of Hereditary Inclusion Body Myopathy and move therapeutic agents towards clinical trials.</p>\",\"PeriodicalId\":73138,\"journal\":{\"name\":\"Gene regulation and systems biology\",\"volume\":\"3 \",\"pages\":\"181-90\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2009-10-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.4137/grsb.s2594\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Gene regulation and systems biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4137/grsb.s2594\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Gene regulation and systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4137/grsb.s2594","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 13
摘要
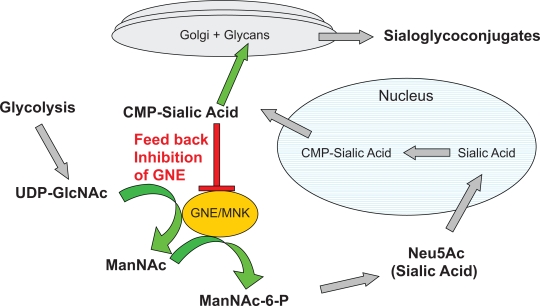
遗传性包涵体肌病2型(HIBM2)是一种以进行性肌肉无力为特征的肌病,成人早期发病。该疾病是葡萄糖胺(udp - n -乙酰)-2- epimase / n -乙酰氨基甘露胺激酶基因(GNE)隐性突变的结果,导致酶功能和唾液酸水平降低。大多数HIBM2患者来自伊朗-犹太人或日本血统,但在世界范围内也发现了孤立病例。本文综述了HIBM2的诊断标准。目前的研究重点是疾病的生物学和GNE在唾液酸途径中的作用。最后,讨论了治疗研究和动物模型,重点讨论了未来的研究,以更好地了解遗传性包涵体肌病的病理,并将治疗药物推向临床试验。
Hereditary inclusion body myopathy type 2 (HIBM2) is a myopathy characterized by progressive muscle weakness with early adult onset. The disease is the result of a recessive mutation in the Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase gene (GNE), which results in reduced enzyme function and sialic acid levels. A majority of individuals with HIBM2 are from Iranian-Jewish or Japanese decent, but isolated cases have been identified world wide. This article reviews the diagnostic criteria for HIBM2. Current research with a highlight on the biology of the disease and the role of GNE in the sialic acid pathway are assessed. Finally, therapeutic investigations and animal models are discussed with a focus on future studies to better understand the pathology of Hereditary Inclusion Body Myopathy and move therapeutic agents towards clinical trials.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
