Hye In Woo, Hyung-Doo Park, Yong-Wha Lee, Dong Hwan Lee, Chang-Seok Ki, Soo-Youn Lee, Jong-Won Kim
{"title":"2例韩国中链酰基辅酶a脱氢酶缺乏症的临床、生化和遗传分析。","authors":"Hye In Woo, Hyung-Doo Park, Yong-Wha Lee, Dong Hwan Lee, Chang-Seok Ki, Soo-Youn Lee, Jong-Won Kim","doi":"10.3343/kjlm.2011.31.1.54","DOIUrl":null,"url":null,"abstract":"<p><p>Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is an autosomal recessive hereditary metabolic disorder of mitochondrial fatty acid β-oxidation. It is characterized by hypoketotic hypoglycemia, hyperammonemia, seizure, coma, and sudden infant death syndrome-like illness. The most frequently isolated mutation in the acyl-CoA dehydrogenase, medium-chain (ACADM) gene of Caucasian patients with MCADD is c.985A>G, but ethnic variations exist in the frequency of this mutation. Here, we describe 2 Korean pediatric cases of MCADD, which was detected during newborn screening by tandem mass spectrometry and confirmed by molecular analysis. The levels of medium-chain acylcarnitines, including octanoylcarnitine (C8), hexanoylcarnitine (C6), and decanoylcarnitine (C10), were typically elevated. Molecular studies revealed that Patient 1 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.461T>G (p.L154W) mutations, and Patient 2 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.1189T>A (p.Y397N) mutations. We detected asymptomatic patients with MCADD by using a newborn screening test and confirmed it by ACADM mutation analysis. This report presents evidence of the biochemical and molecular features of MCADD in Korean patients and, to the best of our knowledge, this is the first report of the c.461T>G mutation in the ACADM gene.</p>","PeriodicalId":17890,"journal":{"name":"Korean Journal of Laboratory Medicine","volume":"31 1","pages":"54-60"},"PeriodicalIF":0.0000,"publicationDate":"2011-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3343/kjlm.2011.31.1.54","citationCount":"14","resultStr":"{\"title\":\"Clinical, biochemical and genetic analyses in two Korean patients with medium-chain acyl-CoA dehydrogenase deficiency.\",\"authors\":\"Hye In Woo, Hyung-Doo Park, Yong-Wha Lee, Dong Hwan Lee, Chang-Seok Ki, Soo-Youn Lee, Jong-Won Kim\",\"doi\":\"10.3343/kjlm.2011.31.1.54\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is an autosomal recessive hereditary metabolic disorder of mitochondrial fatty acid β-oxidation. It is characterized by hypoketotic hypoglycemia, hyperammonemia, seizure, coma, and sudden infant death syndrome-like illness. The most frequently isolated mutation in the acyl-CoA dehydrogenase, medium-chain (ACADM) gene of Caucasian patients with MCADD is c.985A>G, but ethnic variations exist in the frequency of this mutation. Here, we describe 2 Korean pediatric cases of MCADD, which was detected during newborn screening by tandem mass spectrometry and confirmed by molecular analysis. The levels of medium-chain acylcarnitines, including octanoylcarnitine (C8), hexanoylcarnitine (C6), and decanoylcarnitine (C10), were typically elevated. Molecular studies revealed that Patient 1 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.461T>G (p.L154W) mutations, and Patient 2 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.1189T>A (p.Y397N) mutations. We detected asymptomatic patients with MCADD by using a newborn screening test and confirmed it by ACADM mutation analysis. This report presents evidence of the biochemical and molecular features of MCADD in Korean patients and, to the best of our knowledge, this is the first report of the c.461T>G mutation in the ACADM gene.</p>\",\"PeriodicalId\":17890,\"journal\":{\"name\":\"Korean Journal of Laboratory Medicine\",\"volume\":\"31 1\",\"pages\":\"54-60\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2011-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.3343/kjlm.2011.31.1.54\",\"citationCount\":\"14\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Korean Journal of Laboratory Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3343/kjlm.2011.31.1.54\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Korean Journal of Laboratory Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3343/kjlm.2011.31.1.54","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 14
摘要
中链酰基辅酶a脱氢酶缺乏症(MCADD)是一种常染色体隐性遗传性线粒体脂肪酸β氧化代谢疾病。它的特点是低酮性低血糖、高氨血症、癫痫发作、昏迷和婴儿猝死综合征样疾病。高加索MCADD患者ACADM基因中最常见的突变为c.985A>G,但该突变的频率存在种族差异。在这里,我们描述了2例韩国儿童MCADD病例,这是在新生儿筛查中通过串联质谱检测到的,并通过分子分析证实。中链酰基肉碱,包括辛烷基肉碱(C8)、己烷基肉碱(C6)和癸烷基肉碱(C10)的水平明显升高。分子研究表明,患者1为c.449_452delCTGA (p.Thr150ArgfsX4)和c.461T>G (p.L154W)突变的复合杂合子,患者2为c.449_452delCTGA (p.s thr150argfsx4)和c.1189T> a (p.p y397n)突变的复合杂合子。我们通过新生儿筛查试验发现无症状MCADD患者,并通过ACADM突变分析确诊。本报告提供了韩国患者MCADD的生化和分子特征的证据,据我们所知,这是ACADM基因c.461T>G突变的首次报道。
Clinical, biochemical and genetic analyses in two Korean patients with medium-chain acyl-CoA dehydrogenase deficiency.
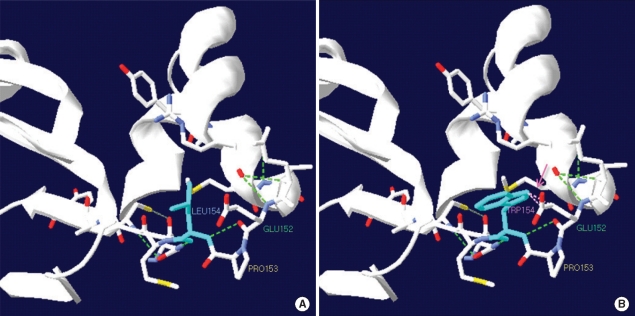
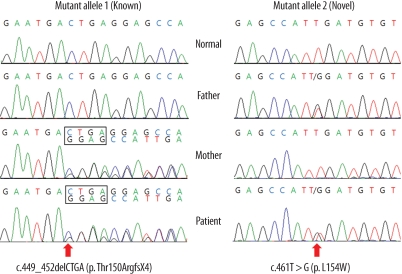
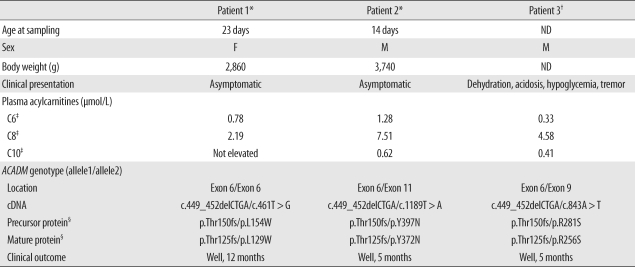
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is an autosomal recessive hereditary metabolic disorder of mitochondrial fatty acid β-oxidation. It is characterized by hypoketotic hypoglycemia, hyperammonemia, seizure, coma, and sudden infant death syndrome-like illness. The most frequently isolated mutation in the acyl-CoA dehydrogenase, medium-chain (ACADM) gene of Caucasian patients with MCADD is c.985A>G, but ethnic variations exist in the frequency of this mutation. Here, we describe 2 Korean pediatric cases of MCADD, which was detected during newborn screening by tandem mass spectrometry and confirmed by molecular analysis. The levels of medium-chain acylcarnitines, including octanoylcarnitine (C8), hexanoylcarnitine (C6), and decanoylcarnitine (C10), were typically elevated. Molecular studies revealed that Patient 1 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.461T>G (p.L154W) mutations, and Patient 2 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.1189T>A (p.Y397N) mutations. We detected asymptomatic patients with MCADD by using a newborn screening test and confirmed it by ACADM mutation analysis. This report presents evidence of the biochemical and molecular features of MCADD in Korean patients and, to the best of our knowledge, this is the first report of the c.461T>G mutation in the ACADM gene.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
